Konzepte zur Überwindung der Blut-Hirn-Schranke
Konzepte zur Überwindung der Blut-Hirn-Schranke ermöglichen es, dem Gehirn für therapeutische Zwecke Wirkstoffe zuzuführen. Die Blut-Hirn-Schranke ist eine dynamische Grenzfläche, die über Influx (Zufluss, wörtlich: Einströmen) und Efflux (Abfluss) kontrolliert, welche Nährstoffe, Arzneistoffe, Drogen, Xenobiotika und sonstige Verbindungen dem Gehirn zugeführt werden können.[1] Dadurch gewährleistet sie dem Zentralnervensystem (ZNS) ein optimales Milieu.

Ihre Schutzfunktion macht die Blut-Hirn-Schranke jedoch auch zu einer Barriere für viele potenzielle Wirkstoffe und vereitelt so deren Einsatz in medikamentösen Therapien. Etwa 98 % der potenziellen Neuropharmaka scheitern daran.[2] So lassen sich nur relativ wenige neurologische und psychiatrische Erkrankungen wie beispielsweise affektive Störungen wie Depressionen, Epilepsie oder chronische Schmerzen mit kleinen lipophilen Wirkstoffen behandeln.[3][4]
Dagegen gibt es keine Therapie für neurodegenerative Erkrankungen wie die Alzheimer-Krankheit, Chorea Huntington und die Amyotrophe Lateralsklerose (ALS).[2] Für Gehirntumoren, Schlaganfälle, Rückenmarksverletzungen und Schädel-Hirn-Traumata sind keine effektiven medikamentösen Therapien bekannt. Auch bei im Kindesalter auftretenden Syndromen wie Autismus, lysosomalen Speicherkrankheiten, dem Fragiles-X-Syndrom oder Ataxie stellt die Blut-Hirn-Schranke eine Barriere dar, die bisherige medikamentöse Therapieansätze verhindert.[5] Selbst bei Erkrankungen wie Multipler Sklerose kann die Progression der Erkrankung im Zentralnervensystem nicht gestoppt werden, da die verabreichten Medikamente nur in der Peripherie wirken. Prinzipiell könnten viele dieser Erkrankungen mit Wirkstoffen, beispielsweise auf Basis von Enzymen, Genen oder biotechnologisch hergestellten Proteinen, behandelt werden – wenn sie die Blut-Hirn-Schranke überwinden könnten. Eine Therapie ist aber nur möglich, wenn diese Substanzen in ausreichender, das heißt therapeutisch wirksamer Konzentration auch an den Wirkort – also das Zentralnervensystem – gelangen können.[6] Es wird daher seit Jahrzehnten intensiv an Methoden geforscht, die einen Wirkstofftransport in das Gehirn unter Umgehung oder – idealerweise selektiver – Öffnung der Blut-Hirn-Schranke ermöglichen sollen.[7][8] Eine Reihe von Strategien zur Überwindung der Blut-Hirn-Schranke wurde dabei entwickelt oder befindet sich noch im Entwicklungsstadium.[9][10]
Umgehen der Blut-Hirn-Schranke – intrathekale und intraventrikuläre Wirkstoffapplikation
Bearbeiten
Die naheliegendste Form des Wirkstofftransportes in das ZNS unter Umgehung der Blut-Hirn-Schranke stellt die Injektion direkt in den Liquor cerebrospinalis (intrathekal) oder direkt in die Hirnventrikel (intraventrikulär)[11] dar. Der Wirkstoff wird dabei direkt in den Liquor injiziert. Angewendet wird dieses Verfahren beispielsweise als intrathekale Chemotherapie[12][13] unter anderem mit dem Folsäure-Antagonisten Methotrexat (MTX), mit Cytarabin (AraC) und Cortisol; speziell bei Patienten mit akuter lymphatischer Leukämie und aggressiven Lymphomen.[14] Die drei Wirkstoffe werden in der triple intrathecal chemotherapy zur Behandlung der Hirnhaut-Leukämie[15] zusammen in den Liquor appliziert.[16]
Die intrathekale Wirkstoffapplikation ist – verglichen mit der intravenösen (systemischen) Gabe von Wirkstoffen – deutlich aufwändiger und für viele Patienten auch unangenehmer. Darüber hinaus bestehen bei derartigen Darreichungsformen aufgrund der deutlich erhöhten Infektions- und Verletzungsgefahr besonders strenge Anforderungen an Hygiene und technische Fertigkeiten des Anwenders. Durch die Injektion von Wirkstoffen mit Depotwirkung (slow release) können die Behandlungsintervalle auf längere Zeiträume – beispielsweise 14-täglich – gestreckt werden.[16] Weniger aufwändig ist die Verwendung eines Ommaya-Reservoirs, das unter die Kopfhaut implantiert wird. Einen ähnlichen Ansatz bieten implantierbare Medikamentenpumpen.[17] Bei schweren Schmerzzuständen kann diese Methode beispielsweise für die Dosierung von Morphin gewählt werden.[18][19] Auch zur Behandlung von Spastiken, beispielsweise bei Multipler Sklerose mit Baclofen, kann der Wirkstoff über eine solche Pumpe intrathekal appliziert werden.[20][21][22] Die Methode wurde erstmals 1984 angewendet[23] und ist seitdem etabliert.[24][25]
Intrathekal applizierte Wirkstoffe werden meist speziell für diese Darreichungsform formuliert. Sie dürfen beispielsweise keine Bakterizide und eine Reihe anderer Hilfsstoffe enthalten, die in intravenös applizierten Medikamenten übliche Zusatzstoffe sind.[26]
Für einige wenige Erkrankungen ermöglicht die intrathekale beziehungsweise die intraventrikuläre Wirkstoffapplikation eine wirksame Therapie. Für die Behandlung von Hirntumoren sind diese beiden Methoden zur Umgehung der Blut-Hirn-Schranke allerdings nicht geeignet. Die Ursache hierfür liegt in der auf nur wenige Millimeter begrenzten Diffusion der Wirkstoffe in das Parenchym des Gehirns.[27][28][29]
Eine experimentell und therapeutisch nutzbare Lücke in der Blut-Hirn-Schranke sind die in das Gehirn eintretenden Hirnnerven. So konnte gezeigt werden, dass beispielsweise Neurotrophine, Neuropeptide, Insulin, Zytokine und sogar DNA, die über die Nase verabreicht wurden, über den Riechnerv in das Zentralnervensystem gelangen können.[30] Ebenso konnte man über diesen Weg erfolgreich Stammzellen in das Gehirn einschleusen.[31]
Überwindung der Blut-Hirn-Schranke für therapeutische Zwecke
BearbeitenEine intakte Blut-Hirn-Schranke ist für jedes Wirbeltier lebensnotwendig. Für viele Wirkstoffe, die außerhalb des Zentralnervensystems ihre Wirkung entfalten sollen, ist die Retention an der Blut-Hirn-Schranke ein wichtiges Kriterium für die Zulassung, um die sonst zu erwartenden teilweise erheblichen Nebenwirkungen, insbesondere bei dauerhafter Einnahme eines Medikaments, sicher ausschließen zu können. Andererseits stellt die Blut-Hirn-Schranke bei der Behandlung neurologischer Erkrankungen für viele Verbindungen eine unüberwindliche Barriere dar.[5][32]
Lipophilisierung
Bearbeiten-
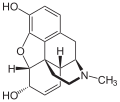 Die Strukturformel von Morphin
Die Strukturformel von Morphin -
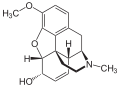 Codein (= 3-Methylmorphin) ist durch eine Methylierung etwas lipophiler als Morphin
Codein (= 3-Methylmorphin) ist durch eine Methylierung etwas lipophiler als Morphin -
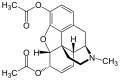 Heroin ist durch zwei Acetylgruppen lipophiler als Morphin, weshalb es wesentlich leichter die Blut-Hirn-Schranke passieren kann.
Heroin ist durch zwei Acetylgruppen lipophiler als Morphin, weshalb es wesentlich leichter die Blut-Hirn-Schranke passieren kann.



Das Diffusionsvermögen eines Moleküls durch die Endothelien der Blut-Hirn-Schranke wird vor allem durch seine Fettlöslichkeit (Lipophilie) und Größe bestimmt. Durch eine Modifizierung des Moleküls mit lipophilen Gruppen kann deshalb eine verbesserte Gehirngängigkeit erreicht werden.[33] Ein klassisches Beispiel hierfür ist die Di-Acetylierung des Naturstoffes Morphin zu Diacetylmorphin (Heroin). Heroin (log P=1,12) zeigt gegenüber Morphin (log P=0,2) eine über 25fach höhere Aufnahme im Gehirn (siehe dazu: Tabelle 1).[34] Entsprechende Ergebnisse werden beim Brain-Uptake-Index (BUI) für radioaktiv markiertes Morphin, Codein und Heroin erhalten, das in die Halsschlagader injiziert wird. Für Morphin liegt der BUI unterhalb der Nachweisgrenze, bei Codein bei 24 % und für Heroin bei 68 %.[35]
Dieses Prodrug-Konzept kann selbst bei peptidischen Wirkstoffen zu einer Verbesserung der Gehirngängigkeit führen.[36]
Das Konzept versagt allerdings bei Molekülen mit einer molaren Masse größer als 500 g·mol−1, da solche Substanzen aufgrund ihrer Größe nicht mehr die Blut-Hirn-Schranke per Diffusion passieren können. Zudem geht mit der Lipophilisierung eine deutlich schlechtere Löslichkeit des Wirkstoffes einher. Bei der oralen Gabe können aber nur gelöste Wirkstoffe im Gastrointestinaltrakt aufgenommen werden. Die Lipophilisierung bewirkt natürlich auch eine erhöhte Aufnahme in anderen, nicht zerebralen, Zellen. Auch gegen Efflux-Transporter, die den eindiffundierten Wirkstoff wieder aus dem Endothel ausschleusen, ist die Lipophilisierung wirkungslos.
Ausnutzung der Transporter
Bearbeiten.svg)

Im Endothel der Blut-Hirn-Schranke sind mehrere Transportsysteme, um das Gehirn mit essentiellen hydrophilen Substanzen zu versorgen. Ein Ansatz, Wirkstoffe in das Gehirn schleusen zu können, ist die Ausnutzung dieser Transporter. Dies wird beispielsweise bei der Therapie der Parkinson-Krankheit angewendet. Daran erkrankte Patienten haben im Gehirn einen Mangel des Neurotransmitters Dopamin. Die Gabe von Dopamin wäre diesbezüglich wirkungslos, da Dopamin die Blut-Hirn-Schranke nicht passieren kann. Verabreicht man dagegen Levodopa, eine nicht-proteinogene α-Aminosäure, so wird diese über den LAT1-Transporter dem Gehirn zugeführt und dort anschließend in Dopamin verstoffwechselt. Der LAT1-Transporter gehört zur Familie der LNAA-Transporter (large neutral amino acid).[37]
Auch das Antiepileptikum Gabapentin, das Antihypertensivum α-Methyldopa und die Zytostatika Melphalan und Acivicin können über LNAA-Transporter die Blut-Hirn-Schranke passieren.[2][38][39][40]
Die Obergrenze für die Ausnutzung der bestehenden Transportsysteme liegt bei einer molaren Masse von etwa 500 bis 600 g·mol−1.[41]
Vektorisierung
BearbeitenEin anderer Weg, um die Blut-Hirn-Schranke mit einem Wirkstoff zu überwinden, ist die Vektorisierung.[42] Dieser Ansatz beruht auf der Beobachtung, dass einige Makromoleküle, wie Transferrin,[43] Low Density Lipoprotein[44] und Insulin[45] über einen mehrstufigen, als rezeptorvermittelte Transzytose bezeichneten Prozess die Blut-Hirn-Schranke überwinden können. Über Rezeptoren, die sich an der Oberfläche der Endothelzellen der Hirnkapillaren befinden und in das Lumen der Blutgefäße hineinragen, werden die Makromoleküle in das Innere der Endothelzellen über Vesikel eingeschleust, um dann auf die andere Seite der Zelle (abluminale Seite) transportiert und ausgeschleust zu werden. Wird ein Wirkstoffmolekül an ein solches Makromolekül gebunden, kann die rezeptorvermittelte Transzytose zur Überwindung der Blut-Hirn-Schranke ausgenutzt werden.
Ein Beispiel hierfür ist der Transferrinrezeptor, der mit Hilfe gegen ihn gerichteter monoklonaler Antikörper zum Transport von Wirkstoffen durch die Blut-Hirn-Schranke genutzt werden kann. Dieser Rezeptor ist gewöhnlicherweise für den Transport von Eisen durch die Blut-Hirn-Schranke zuständig.[46][47] Ein anderes Target ist der Insulinrezeptor, der auch von den Endothelzellen der Blut-Hirn-Schranke exprimiert wird.[48] Mit beiden Vektoren wurden im Tiermodell verschiedene, auch größere, Peptide erfolgreich über die Blut-Hirn-Schranke geschleust.[49] Speziell für die Therapie von neurodegenerativen Erkrankungen, für die nur geringe Wirkstoffkonzentrationen notwendig sind, ist die Vektorisierung ein vielversprechender Ansatz.[50][51] Auch Zytostatika wie beispielsweise Doxorubicin wurden an Transferrinrezeptor-Antikörper gebunden.[52]
Das Phänomen der Transzytose ist jedoch nicht auf Makromoleküle beschränkt. Wenngleich der genaue Mechanismus nicht immer geklärt ist, so konnte gezeigt werden, dass auch kleine Peptide und niedermolekulare Substanzen auf diese Weise in die Zelle gelangen und diese passieren können. Eine Vektorisierung zum Zweck der Passage der Blut-Hirn-Schranke ist somit auch mit kurzen Peptidsequenzen möglich. Als Vektoren für Wirkstoffe, wie beispielsweise Doxorubicin, fanden unter anderem basische Protegrin-Abkömmlinge, wie beispielsweise Syn-B,[53][54] und das aus der Homöodomäne von Antennapedia, einem Transkriptionsfaktor von Drosophila, abgeleitete Penetratin[55] Anwendung. Ein anderer Peptid-Vektor ist das aus elf überwiegend basischen Aminosäuren bestehende und aus der Transduktionsdomäne des HI-Virus isolierte HIV-TAT (engl. Trans-Activator of Transcription).[51][56] Ein Peptid mit ähnlichen Eigenschaften ist das aus 27 Aminosäuren aufgebaute Transportan, ein zellpenetrierendes Peptid.[57]
Mit transgenen Makrophagen können Proteine durch die Blut-Hirn-Schranke geschleust werden.[58][59][60]
Kationisierung
BearbeitenPositiv geladene Moleküle (Kationen) können mit Hilfe der adsorptionsvermittelten Transzytose, auch kationischer Transport genannt, die Blut-Hirn-Schranke überwinden.[61] Bei der adsorptionsvermittelten Transzytose bewirken elektrostatische Wechselwirkungen zwischen der durch Glykoproteine negativ geladenen Zelloberfläche und positiv geladenen Molekülen eine unspezifische Bindung an die Oberfläche von Zellen, in deren Folge eine Aufnahme und ein Transport durch das Zytoplasma der Endothelien erfolgt.[62] Die kationische Transzytose durch das Endothel der Blut-Hirn-Schranke ermöglicht einen höheren Grad des Stofftransportes als die rezeptorvermittelte Transzytose.[63]
Die Kationisierung von Antikörpern wurde in einer Reihe unterschiedlicher Studien und Anwendungsfeldern erfolgreich zur Passage der Blut-Hirn-Schranke eingesetzt. So beispielsweise, um β-Amyloidplaques sichtbar zu machen[64][65] oder Mitochondrien zu targetieren.[66]
Eine positive Ladung weisen bereits Peptide und Proteine auf, deren isoelektrischer Punkt im Basischen liegt.[51] Ein Ansatz, die Aufnahme nicht basischer Peptide und Proteine im Gehirn zu verbessern, ist, diese mit Hilfe von natürlich vorkommenden Polyaminen, wie beispielsweise Putrescin, Spermidin oder Spermin, chemisch zu modifizieren.[67][68] Eine Alternative dazu ist die im Kapitel Vektorisierung beschriebene Konjugation von Wirkstoffpeptiden und -proteinen an basische Peptide wie Syn-B.[61] Auch synthetische Polyamine, wie beispielsweise Polyethylenimin, können zum erleichterten Transport von Wirkstoffen und DNA durch die Blut-Hirn-Schranke eingesetzt werden.[69]
Der Effekt der Kationisierung ermöglicht zwar die Passage von Wirkstoffen und Diagnostika über die Blut-Hirn-Schranke, bewirkt aber gleichzeitig eine erheblich gesteigerte Aufnahme der applizierten Dosis in Leber und Nieren – mit den entsprechenden zu erwartenden Nebenwirkungen.
Nanopartikel
Bearbeiten


In den 1990er Jahren wurde in Versuchen mit Nanopartikeln, die aus biokompatiblen Polymeren aufgebaut sind, festgestellt, dass diese Partikel unter bestimmten Umständen in der Lage sind, die Blut-Hirn-Schranke zu passieren. Der Durchmesser dieser Partikel liegt üblicherweise bei 50 bis 300 nm. Die unfunktionalisierten, reinen Polymerpartikel sind in dieser Form nicht in der Lage durch das Endothel zum Gehirn transportiert zu werden. Der rezeptorvermittelte Transport ist nur durch eine spezielle Funktionalisierung, meist mit Polysorbat 80 oder Poloxameren,[70] möglich. Als Polymere werden meist Polylactide (PLA), Polylactid-co-Glycolid (PLGA) und verschiedene Polycyanoacrylate, wie beispielsweise Polybutylcyanoacrylat (PBCA),[71] verwendet, die pharmakologisch unbedenklich sind und für andere Anwendungen, beispielsweise als chirurgisches Nähmaterial, zugelassen sind. In die Partikel eingeschlossene Wirkstoffe können mittels rezeptorvermittelter Transzytose zum Gehirn transportiert werden.[72]
Die wesentlichen Voraussetzungen für die Hirngängigkeit der Nanopartikel ist – neben ihrer Größe – eine möglichst lange Zirkulationszeit im Blut und die passende Oberflächencharakteristik. Die Plasmahalbwertszeit wird meist durch eine PEGylierung erreicht und die Wechselwirkung am Endothel mit dem bereits beschriebenen Polysorbat.[73] Der genaue Transportmechanismus ist noch nicht endgültig geklärt. Der Polysorbat-Überzug der Partikel führt aber offensichtlich im Blutplasma zu einer Adsorption von Apolipoprotein E oder B an die Partikel. Dadurch werden die Nanopartikel als LDL-Mimetikum vom LDL-Rezeptor erkannt und in das Innere des Endothels transportiert. Danach wird der Wirkstoff entweder im Endothel freigesetzt, wodurch er per Diffusion zum Gehirn gelangen kann, oder die Partikel werden vollständig durch die abluminale Seite zum Gehirn ausgeschleust (Transzytose).[74]
Der nanopartikuläre Wirkstofftransport ist derzeit noch in der präklinischen Forschung. Im Tiermodell (Ratte) wurden vielversprechende Ergebnisse bei der Behandlung von transplantierten Glioblastomen erzielt. Dabei wurden die Partikel mit Doxorubicin beladen.[75] Der Transport von Doxorubicin in das Gehirn konnte dabei um den Faktor 60 gesteigert werden.[76] Die wegen der weitgehenden Undurchlässigkeit der Blut-Hirn-Schranke für Chemotherapeutika nur schwer zu realisierende Chemotherapie bei Gehirntumoren ist eines der Hauptziele bei der Entwicklung dieser nanopartikulären Wirkstoff-Träger-Systeme.[77]
Mit speziellen Liganden ist darüber hinaus die gewebe- beziehungsweise rezeptorspezifische Targetierung der Nanopartikel denkbar.[78]
Neben dem nanopartikulären Ansatz mit Polymeren sind auch nanoskalige Liposomen[79][80] und Dendrimere als potenzielle Wirkstofftransporter in der präklinischen Erprobung.[81] Besondere Beachtung findet dabei auch die im Rahmen der gesamten Nanotechnologie stattfindende Diskussion über ihre Risiken.[82]
Lösungsmittel und Tenside
BearbeitenIntravenös applizierte Verbindungen, wie Ethanol, Dimethylsulfoxid[83] oder Glycerin, können zu einer lösungsmittelinduzierten Öffnung der Blut-Hirn-Schranke führen. Im Tiermodell (Küken) liegt dabei die Konzentration an Lösungsmittel oberhalb von 1 mg pro kg Körpergewicht.[84] Diese Verbindungen stören vermutlich die Funktion der Zellmembran im Endothel, wodurch der Stofftransport durch transzelluläre Diffusion ermöglicht wird.[5]

Werden kurzkettige Alkylglycerole, wie beispielsweise 1-O-Hexyldiglycerol, zusammen mit Marker-Substanzen in die Halsschlagader von Mäusen oder Ratten injiziert, so erhöht sich die Aufnahme dieser Marker im Gehirn signifikant. Größere Moleküle, die sonst nicht die Blut-Hirn-Schranke passieren, wie beispielsweise Methotrexat, Vancomycin oder Gentamicin, können – bedingt durch die Anwesenheit des Alkylglycerols – in das Gehirn diffundieren.[85] Dieser Effekt wird bei der intravenösen Gabe von Alkylglycerol nicht beobachtet. Die amphipathischen Glycerole öffnen die Blut-Hirn-Schranke dabei für ungefähr 5 bis 120 Minuten.[86] Die Konzentrationen der Alkylglycerole liegen im millimolaren Bereich. Offensichtlich bilden diese tensidähnlichen Verbindungen mit den Wirkstoffen, beziehungsweise Markern, vesikuläre Strukturen.[87] Alkylglycerole sind weitgehend untoxisch und pharmakologisch unbedenklich.[88][89] Der Mechanismus der Überwindung der Blut-Hirn-Schranke ist größtenteils noch ungeklärt. Es handelt sich aber offensichtlich um einen Transport durch die Tight Junctions.[86]
Auch das Tensid Natriumlaurylsulfat erhöht bei der Injektion in die Halsschlagader die Durchlässigkeit der Blut-Hirn-Schranke deutlich.[90] Natriumlaurylsulfat ist ein pharmakologischer Hilfsstoff, der in verschiedenen Wirkstoffformulierungen zur Anwendung kommt. Die entsprechende Applikation solcher Formulierungen kann daher zu unerwarteten Ergebnissen führen. So bewirkte der Hilfsstoff Natriumlaurylsulfat in einer Formulierung mit Interleukin-2, dass die Blut-Hirn-Schranke bei Katzen für die Markersubstanz Meerrettichperoxidase überraschend durchlässig wurde.[84][91] Ähnliche Effekte wurden auch mit dem Hilfsstoff Polysorbat-80 beobachtet. Hierzu genügen bei einer Maus schon Dosen im Bereich von 3 mg pro kg Körpergewicht.[92] Kyotorphin, ein neurophysiologisch aktives Dipeptid, ist nicht in der Lage die Blut-Hirn-Schranke zu passieren und eine neurologische Wirkung zu zeigen. Nur in Verbindung mit Polysorbat-80 wird die neurologische Wirkung erreicht.[84][93]
Efflux-Inhibierung
Bearbeiten

Viele Moleküle sind sowohl wegen ihrer Größe als auch ihrer Lipophilie in der Lage die Blut-Hirn-Schranke zu passieren. Sie werden aber nach dem Diffundieren in das Zytoplasma der Endothelien durch Efflux-Pumpen, wie beispielsweise P-Glykoprotein, wieder zurück in das Lumen transportiert. Eine Strategie, um diese Moleküle dennoch dem Gehirn zugänglich zu machen, ist das Ausschalten dieser Efflux-Transporter. Prinzipiell ist dies möglich durch:
- Genregulation in der transkriptionalen oder translationalen Phase
- Veränderungen der Membran-Targetierung nach der Synthese der Transporter in den Ribosomen
- Unterbinden des Transportes durch Inhibitoren (Co-Drugs)
Während die ersten beiden Methoden sich noch in einem sehr frühen Entwicklungsstadium auf der Ebene von Zellkulturen befinden, liegen bei den Efflux-Inhibitoren ausgiebige Erfahrungen am Tier und aus klinischen Studien am Menschen vor.[94]
Mittlerweile ist eine Reihe von Substanzen bekannt, die den Efflux – speziell durch P-Glykoprotein – inhibieren.[95][96]
Mäuse, bei denen das MDR1-Gen abgeschaltet (Knockout) wurde, so dass im Endothel kein P-Glykoprotein produziert wird, zeigen für eine Reihe von Wirkstoffen eine signifikant erhöhte Aufnahme im Gehirn über die Blut-Hirn-Schranke. Im Vergleich zum Wildtyp der Maus stieg beispielsweise das Konzentrationsverhältnis Gehirn zu Blut bei den HIV-Protease-Inhibitoren Nelfinavir, Indinavir und Saquinavir um den Faktor 7 bis 36 an.[97] Bei den Taxanen Docetaxel und Paclitaxel erhöht sich die Konzentration im Gehirn um den Faktor 7 bis 28[98][99][100] und bei Digoxin um den Faktor 10.[101] Bei Verapamil wird die Aufnahme im Gehirn um den Faktor 8,5 verbessert.[102]
Bei Wildtypen von Mäusen und Ratten, denen selektiv wirkende P-Glykoprotein-Inhibitoren, wie beispielsweise Valspodar (PSC 833, ein Ciclosporin-Derivat), Elacridar (GF120918) und Zosuquidar (LY335979),[99][100][101][103] verabreicht wurden, konnten vergleichbare Ergebnisse erhalten werden. Bei Ratten, denen Ciclosporin verabreicht wurde, erhöht sich die Konzentration von Verapamil im Gehirn um den Faktor 9,6.[102][104]
Verapamil – ein als Calciumantagonist zugelassenes Arzneimittel – ist im Tierversuch selbst ein wirksames Co-Drug, das die Aufnahme bei nachfolgend applizierten Wirkstoffen im Gehirn deutlich erhöhen kann. Dies wurde im Tiermodell unter anderem bei zytostatischen Vincaalkaloiden nachgewiesen.[105][106] Eine ähnliche Wirkung zeigen Procyanidine.[107]
Nachteilig bei dem Ansatz der Efflux-Inhibierung ist, dass die verabreichten Inhibitoren – speziell der ersten Generation, wie Verapamil und Ciclosporin – selbst pharmakologisch aktiv sind und so eine Reihe von unerwünschten Nebenwirkungen haben. Bei der zweiten und dritten Generation von P-Glykoprotein-Inhibitoren sind diese Effekte deutlich reduziert.[94] Außerdem wird bei allen Zellen – die P-Glykoprotein exprimieren – selbiges inhibiert. So sind bei der systemischen Gabe von Efflux-Inhibitoren auch die apikale Seite der Darm-Epithelien, der Gallenkanälchen (Bilis canaliculi), der Nierenkanälchen und der Plazenta, sowie an der luminalen Seite die der Hodenkanälchen betroffen.[108]
BCRP (Brustkrebs-Resistenz-Protein, Breast Cancer Resistance Proteine), der zweitwichtigste Efflux-Transporter der Blut-Hirn-Schranke, hat offensichtlich kaum einen Einfluss auf den Transport von Wirkstoffen.[94] Dies wurde bei Versuchen an Knockout-Mäusen festgestellt, bei denen das BCRP-codierende ABCG2-Gen abgeschaltet wurde.[109]
Die Efflux-Inhibierung wird insbesondere in der Krebstherapie verfolgt, da viele Krebszellen im Therapieverlauf P-Glykoprotein stark exprimieren und sich dadurch der Wirkung von Zytostatika weitgehend entziehen können. Die Tumoren sprechen dann nicht mehr auf die verabreichten Zytostatika an.[110][111][112]
Öffnen der Blut-Hirn-Schranke für therapeutische Zwecke
Bearbeiten
Das Öffnen der Blut-Hirn-Schranke für therapeutische Zwecke ist, neben den beiden zuvor gezeigten Prinzipien, eine weitere Strategie, um Wirkstoffe dem Gehirn zuzuführen, die normalerweise nicht in der Lage sind die Blut-Hirn-Schranke zu passieren. Das Ziel dieser Verfahren ist eine möglichst reversible Öffnung oder zumindest Lockerung der Tight Junctions, um einen parazellulären Wirkstofftransport in das Gehirn zu ermöglichen. Mit dem zunehmenden Verständnis des molekularen Aufbaus der Blut-Hirn-Schranke – und hierbei vor allem der Tight Junctions – wurden neue Wege und Verfahren zur pharmakologischen, aber auch physikalischen, Öffnung der Blut-Hirn-Schranke entwickelt.[113] Die meisten dieser Verfahren befinden sich noch in der präklinischen Erprobung.
Beim Öffnen der Blut-Hirn-Schranke besteht allgemein die Gefahr, dass für das Gehirn toxische Plasmaproteine eindiffundieren und dann chronische Neuropathologien auslösen können.[114]
Tight-Junction-Modulation
BearbeitenVerbindungen, die einen Einfluss auf die Tight Junctions haben, werden als Tight-Junction-Modulatoren bezeichnet. Durch die Fortschritte im Bereich der genomischen Wirkstoffentwicklung, des High-Throughput Screening, der kombinatorischen Chemie und der Bioinformatik, wurde eine Reihe von Substanzen entwickelt beziehungsweise identifiziert, die in der Lage sind unmittelbar die einzelnen Peptide der Tight Junctions und Adherens Junction zu targetieren und damit den Zell-Zell-Kontakt der Endothelien zu modulieren.[115][116]
Modulatoren, die unmittelbar die Tight Junctions targetieren, leiten sich beispielsweise von den Enterotoxinen der Bakterien Vibrio cholerae und Clostridium perfringens ab. Vibrio cholerae – ein Cholera-Erreger – bildet unter anderem das Zonula-Occludens-Toxin (ZOT, Zonula occludens = Tight Junction). ZOT ist ein aus 399 Aminosäuren aufgebautes, 45 kDa schweres Protein, das im Darm mit einem Oberflächenrezeptor – dem ZOT-Rezeptor – der dortigen Endothelien interagiert und dadurch eine intrazelluläre Signalkaskade auslöst, die noch nicht vollständig aufgeklärt ist. Es wird unter anderem das Enzym Proteinkinase A aktiviert, das den Abbau der Tight Junctions katalysiert.[117][118] An Einzellagen zerebraler Endothelien bewirkt ZOT in vitro eine deutliche Reduzierung des transendothelialen elektrischen Widerstandes (TEER), die reversibel ist. Für die Markermoleküle Saccharose, Inulin, Paxlitaxel und Doxorubicin wird die parazelluläre Permeabilität signifikant erhöht.[119] Auch das 12 kDa schwere aktive ZOT-Fragment ΔG sowie die aus nur sechs Aminosäuren (im Einbuchstabencode: FCIGRL) bestehende aktive ZOT-Domäne (AT1002) binden an den ZOT-Rezeptor.[113][120]
Das aus 44 Aminosäuren bestehende OCC2-Peptid bindet selektiv an die zweite Domäne des Tight-Junction-Proteins Occludin, wodurch ebenfalls der parazelluläre Transport erleichtert wird.[121]
Bradykinin, ein aus neun Aminosäuren aufgebautes gefäßerweiternd wirkendes Oligopeptid, bindet an die B2-Rezeptoren der luminalen Seite der Endothelien. Als Folge davon steigt die Konzentration an freien intrazellulären Calcium-Ionen und der mit den transmembranen Tight-Junction-Proteinen Occludin und Claudin verbundene Aktin-Myosin-Komplex wird aktiviert, wodurch die Tight Junctions geöffnet werden.[7][122][123]
Osmotische Öffnung der Blut-Hirn-Schranke
Bearbeiten
Kurz nach der Entdeckung der Tight Junctions wurde 1970 die These aufgestellt, dass die Einwirkung von hyperosmotischen Lösungen auf die Endothelzellen die Blut-Hirn-Schranke öffnen könne.[124] 1980 wurde diese Methode erstmals angewendet[125] und 1984 wurde durch elektronenmikroskopische Aufnahmen der experimentelle Beweis für diese These erbracht. Elektronendichte Marker waren durch die Tight Junctions in das Gehirn diffundiert.[126]
Über die Arteria carotis interna werden hyperosmolare Lösungen, beispielsweise von Mannitol oder Arabinose infundiert. Der unterschiedliche osmotische Druck zwischen den Endothelzellen und der infundierten Lösung bewirkt einen Flüssigkeitsverlust in den Endothelzellen, der zu deren Schrumpfung führt. Durch die Schrumpfung entstehen Zugkräfte zwischen den Zellen, was zu einer Öffnung der Tight Junctions und somit zur Öffnung der Blut-Hirn-Schranke führt.[127][128]
Aufgrund des Konzentrationsgradienten zwischen intravasalem und interstitiellem Raum fließt in größerer Menge Wasser aus dem Plasma ins Gehirn zurück (bulk flow). Dadurch werden im Wasser gelöste Moleküle in das Gehirn eingeschwemmt, wobei ein Ödem entsteht.[125][129][130][131][132]
Die durch die Schrumpfung der Endothelzellen bewirkte Öffnung der Tight Junctions beträgt etwa 20 nm.[132] Dadurch können Moleküle mit einem hydrodynamischem Durchmesser von ebenfalls etwa 20 nm in das Gehirn eindiffundieren.[133] Die Öffnung der Blut-Hirn-Schranke ist bei dieser Methode reversibel. Zehn Minuten bis spätestens zwei Stunden nach der Infundierung ist sie wieder vollständig hergestellt.[123][134] Die Einwirkungszeit der hyperosmolaren Lösung beträgt etwa 30 Sekunden. Durch eine Vorbehandlung mit einem Na+/Ca2+-Kanalblocker kann die Öffnungsdauer der Blut-Hirn-Schranke verlängert werden.
Das Verfahren wurde im Tiermodell mit einer Vielzahl von wasserlöslichen Wirkstoffen, Peptiden, Antikörpern, Enzymen und viralen Vektoren für die Gentherapie getestet. Eine Reihe von klinischen Studien zur Therapie von Gehirntumoren in Kombination mit Chemotherapeutika werden in verschiedenen Kliniken durchgeführt.[135] Die Ergebnisse sind für diese Anwendung vielversprechend.[136]
Ultraschall
Bearbeiten





Die Blut-Hirn-Schranke lässt sich durch fokussierten Ultraschall öffnen. Dieser Effekt wurde erstmals 1956 nachgewiesen. Die Öffnung der Blut-Hirn-Schranke konnte durch die Anfärbung des Gehirns mit Trypanblau – einem Vitalfarbstoff, der normalerweise die Blut-Hirn-Schranke nicht passieren kann – und durch radioaktiv markiertes Phosphat nachgewiesen werden. Mikroskopisch konnten keine Veränderungen am Endothel beobachtet werden. Die Anwendung des Ultraschalls führte allerdings zu Hirnverletzungen.[137] 1960 wurde dann erstmals die Blut-Hirn-Schranke mit nur einer geringen Schädigung des umliegenden Parenchyms durch Ultraschall geöffnet.[138] Alle diese Versuche wurden mit hochintensivem fokussiertem Ultraschall, mit Leistungen im Bereich von 4000 Watt/cm², durchgeführt. Dabei entstehen Kavitationsblasen, die das Gewebe irreversibel zerstören können.[6]
- Fokussierender Ultraschall mit Mikrobläschen
Die Öffnung der Blut-Hirn-Schranke mit Ultraschall und gleichzeitig applizierten Mikrobläschen (Microbubbles) kam 2001 zum ersten Mal zur Anwendung.[139] Der Ansatz dabei ist, dass keine Kavitationsblasen generiert werden müssen, sondern injizierte Mikrobläschen die Funktion der sonst durch die hohe Ultraschallleistung erzeugten Kavitationsblasen übernehmen. Dadurch kann die Leistung des Ultraschalls deutlich reduziert werden; es besteht keine Gefahr mehr den behandelten Schädel, beziehungsweise das umliegende Gewebe, zu überhitzen. Die Technik ist mittlerweile so weit entwickelt, dass bei der Öffnung der Blut-Hirn-Schranke keine Apoptose, keine Ischämie oder sonstige Langzeitschädigung im Gehirn nachzuweisen sind. Wenige Stunden nach der Behandlung ist der alte Zustand der Blut-Hirn-Schranke wiederhergestellt.[6]
Der Fokus des Ultraschalls kann auf beliebige Areale im Gehirn gerichtet werden. Dadurch kann die Blut-Hirn-Schranke selektiv, auf bestimmte Hirnareale begrenzt, geöffnet werden. So können applizierte Wirkstoffe gezielt in diese Areale diffundieren.[140] Die behandelten Areale lassen sich durch eine simultan laufende Magnetresonanztomographie (MRT) genau verfolgen. Dabei dringt das für die MRT verwendete Kontrastmittel, beispielsweise Gadopentetat-Dimeglumin, nur durch die geöffneten Areale der Blut-Hirn-Schranke in das Gehirn ein. Diese Bereiche werden dadurch im MRT deutlich sichtbar hervorgehoben. Das hochpolare Gadopentetat-Dimeglumin ist nicht in der Lage die ungeöffneten Bereiche der Blut-Hirn-Schranke zu passieren.
Im Tiermodell Maus werden bei der Anwendung von fokussiertem Ultraschall mit Mikrobläschen Frequenzen im Bereich von 0,5 und 2 MHz[141] mit kurzen Pulslängen im Millisekundenbereich und Wiederholfrequenzen im Bereich von 1 Hz, über einen Zeitraum von weniger als einer Minute angewendet.[142] Der optimale Frequenzbereich liegt unterhalb von 1 MHz.[143] Die akustische Leistung beträgt weniger als ein Watt. Die verwendeten Mikrobläschen sind meist zugelassene Kontrastmittel aus der kontrastmittelverstärkten Sonographie. Sie haben typischerweise einen Durchmesser von 3 bis 4,5 µm, bestehen beispielsweise aus Humanalbumin und sind mit Perfluorpropan oder ähnlichen Schwergasen gefüllt.[144]
- Mechanismus
Der Mechanismus zur Öffnung der Blut-Hirn-Schranke durch die Anwendung von fokussiertem Ultraschall, zusammen mit Mikrobläschen, ist noch nicht vollständig aufgeklärt. Die Wechselwirkung von Ultraschall und Mikrobläschen spielt dabei eine große Rolle und führt in vivo zu einer Reihe von biologischen Effekten.[145] Eine wesentliche Rolle scheinen dabei Scherkräfte zu spielen, die durch Mikroströmungen erzeugt werden. Diese Mikroströmungen selbst kommen von Oszillationen der Mikrobläschen im Ultraschallfeld.[145] Von den Endothelien selbst ist wiederum bekannt, dass sie auf Scherkräfte dynamisch reagieren können und Scherkräfte eine kritische Größe für die Homöostase sind.[146] Elektronenmikroskopische Aufnahmen von Kapillargefäßen so behandelter Versuchstiere zeigen sowohl einen transzellulären als auch einen parazellulären Transport von entsprechenden Markermolekülen (Meerrettichperoxidase). Bei dem transzellulären Transport handelt es sich im Wesentlichen um Transzytose. Der parazelluläre Transport wird durch einen komplexen Desintegrationsprozess initiiert, bei dem die Tight Junctions ihre Funktion verlieren.[147]
Die so geöffnete Blut-Hirn-Schranke ist durchlässig für niedermolekulare Chemotherapeutika, wie beispielsweise Doxorubicin[148] und Antikörper, wie Trastuzumab.[149][150][151] Auch die prinzipielle Machbarkeit des Transports von Genen in das Gehirn wurde mit dieser Methode im Tiermodell nachgewiesen.[144][152] Das Verfahren zur Öffnung der Blut-Hirn-Schranke mit Ultraschall und gleichzeitig applizierten Mikrobläschen ist noch ein sehr junges Verfahren. Bisher wurde es nur an Versuchstieren erprobt. Bis zu einer möglichen Zulassung des Verfahrens am Menschen vergehen erfahrungsgemäß noch viele Jahre.
Die für die Bildgebung in der Diagnostik verwendete nicht-fokussierte Ultraschallstrahlung (Sonographie) beeinflusst die Integrität der Blut-Hirn-Schranke – auch bei der Gabe von Kontrastmitteln – nicht.[153]
Literatur
Bearbeiten- A. G. De Boer, W. Sutanto: Drug Transport Across the Blood-brain Barrier. CRC Press, 1997, ISBN 90-5702-032-7
- D. J. Begley u. a.: The Blood-brain Barrier and Drug Delivery to the CNS. Informa Health Care, 2000, ISBN 0-8247-0394-4
- P. Ramge: Untersuchungen zur Überwindung der Blut-Hirn-Schranke mit Hilfe von Nanopartikeln. Shaker Verlag, 1999, ISBN 3-8265-4974-0
Weblinks
BearbeitenEinzelnachweise
Bearbeiten- ↑ A. Tsuji: Small Molecular Drug Transfer across the Blood-Brain Barrier via Carrier-Mediated Transport Systems. In: NeuroRx 2, 2005, S. 54–62. PMID 15717057 (Review).
- ↑ a b c W. M. Pardridge: Blood-brain barrier drug targeting: the future of brain drug development. In: Mol Interv 3, 2003, S. 90–105. PMID 14993430 (Review).
- ↑ A. Ajay u. a.: Designing libraries with CNS activity. In: J Med Chem 42, 1999, S. 4942–4951. PMID 10585204.
- ↑ A. K. Ghose: A knowledgebased approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. In: J Comb Chem 1, 1999, S. 55–68. PMID 10746014.
- ↑ a b c W. W. Pardridge: The blood-brain barrier: bottleneck in brain drug development. In: NeuroRx 2, 2005, S. 3–14. PMID 15717053 (Review).
- ↑ a b c N. Vykhodtseva u. a.: Progress and problems in the application of focused ultrasound for blood-brain barrier disruption. In: Ultrasonics, 48, 2008, S. 279–296. PMID 18511095.
- ↑ a b D. J. Begley: Delivery of therapeutic agents to the central nervous system: the problems and the possibilities. In: Pharmacol Ther, 104, 2004, S. 29–45. PMID 15500907 (Review).
- ↑ W. M. Pardridge: Why is the global CNS pharmaceutical market so under-penetrated? In: Drug Discovery Today, 7, 2002, S. 5–7. PMID 11790589.
- ↑ A. G. de Boer, P. J. Gaillard: Strategies to improve drug delivery across the blood-brain barrier. In: Clin Pharmacokinet, 46, 2007, S. 553–576. PMID 17596102 (Review).
- ↑ A. G. de Boer und P. J. Gaillard: Drug targeting to the brain. In: Annu Rev Pharmacol Toxicol, 47, 2007, S. 323–355. PMID 16961459 (Review).
- ↑ G. Fleischhack u. a.: Pharmacokinetics following intraventricular administration of chemotherapy in patients with neoplastic meningitis. In: Clin. Pharmacokinetics 44, 2005, S. 1–31. PMID 15634030.
- ↑ J. Z. Kerr u. a.: Intrathecal chemotherapy. In: Crit Rev Oncol Hematol, 37, 2001, S. 227–236. PMID 11248578.
- ↑ S. Stapleton, S. Blaney: New agents for intrathecal administration. In: Cancer Invest, 24, 2006, S. 528–534. PMID 16939963.
- ↑ Y. L. Kwong u. a.: Intrathecal chemotherapy for hematologic malignancies: drugs and toxicities. In: Ann Hematol, 88, 2009, S. 193–201.PMID 19050889.
- ↑ S. L. Berg, M. C. Chamberlain: Current treatment of leptomeningeal metastases: systemic chemotherapy, intrathecal chemotherapy and symptom management. In: Cancer Treat Res, 125, 2005, S. 121–146. PMID 16211887.
- ↑ a b A. Ruggiero u. a.: Intrathecal chemotherapy with antineoplastic agents in children. In: Paediatr Drugs, 3, 2001, S. 237–246. PMID 11354696.
- ↑ H. Schneider: Implantierbare Medikamentenpumpen. ( des vom 4. März 2016 im Internet Archive) Info: Der Archivlink wurde automatisch eingesetzt und noch nicht geprüft. Bitte prüfe Original- und Archivlink gemäß Anleitung und entferne dann diesen Hinweis. (PDF) In: Implantatekatalog Teil III (Medizinischer Dienst der Spitzenverbände der Krankenkassen e. V.) April 2000.
- ↑ E. S. Krames: Intrathecal Infusional Therapies for Intractable Pain: Patient Management Guidelines. In: J Pain Symptom Manage, 8, 1993, S. 36–46. PMID 8482892.
- ↑ V. L. Ghafoor u. a.: Intrathecal drug therapy for long-term pain management. In: Am J Health Syst Pharm, 64, 2007, S. 2447–2461. PMID 18029950.
- ↑ G. Ochs u. a.: [Intrathecal baclofen for long-term treatment of spasticity: a multi-centre study. ( des vom 30. Mai 2018 im Internet Archive) Info: Der Archivlink wurde automatisch eingesetzt und noch nicht geprüft. Bitte prüfe Original- und Archivlink gemäß Anleitung und entferne dann diesen Hinweis. Intrathecal baclofen for long-term treatment of spasticity: a multi-centre study.] In: J Neurol Neurosurg Psychiatry, 52, 1989, S. 933–939. PMID 2487035.
- ↑ G. Ochs: Therapeutische Möglichkeiten von Wachstumsfaktoren bei neuromuskulären Erkrankungen. ( des vom 21. November 2010 im Internet Archive) Info: Der Archivlink wurde automatisch eingesetzt und noch nicht geprüft. Bitte prüfe Original- und Archivlink gemäß Anleitung und entferne dann diesen Hinweis. abgerufen am 15. Januar 2008.
- ↑ P. M. Brennan, I. R. Whittle: Intrathecal baclofen therapy for neurological disorders: a sound knowledge base but many challenges remain. In: Br J Neurosurg, 22, 2008, S. 508–519. PMID 18649160.
- ↑ R. D. Penn: Intrathecal Baclofen alleviates spinal cord spacicity. In: The Lancet, 8385, 1985, S. 1078. PMID 6144008.
- ↑ K. S. Lewis, W. M. Mueller: Intrathecal baclofen for severe spasticity secondary to spinal cord injury. In: Ann Pharmacother, 27, 1993, S. 767–774. PMID 8329801.
- ↑ A. Dario und G. Tomei: A benefit-risk assessment of baclofen in severe spinal spasticity. In: Drug Saf, 27, 2004, S. 799–818. PMID 15350152 (Review).
- ↑ Y. W. Cheung u. a.: Stability of cytarabine, methotrexate sodium, and hydrocortisone sodium succinate admixtures. In: Am J Hosp Pharm, 41, 1984, S. 1802–1806. PMID 6496516 (Review).
- ↑ Q. Yan u. a.: Distribution of intracerebral ventricularly administered neurotrophins in rat brain and its correlation with trk receptor expression. In: Exp Neurol, 127, 1994, S. 23–36. PMID 8200435.
- ↑ W. M. Pardridge: CNS drug design based on principles of blood-brain barrier transport. In: J Neurochem, 70, 1998, S. 1781–1792. PMID 9572261 (Review).
- ↑ E. M. Kemper u. a.: Modulation of the blood-brain barrier in oncology: therapeutic opportunities for the treatment of brain tumours? In: Cancer Treat Rev 30, 2004, S. 415–423. PMID 15245774 (Review).
- ↑ Hanson LR, Frey WH: Intranasal delivery bypasses the blood-brain barrier to target therapeutic agents to the central nervous system and treat neurodegenerative disease. In: BMC Neurosci. 9 Suppl 3. Jahrgang, 2008, S. S5, doi:10.1186/1471-2202-9-S3-S5, PMID 19091002, PMC 2604883 (freier Volltext).
- ↑ Danielyan L, Schäfer R, von Ameln-Mayerhofer A, et al.: Intranasal delivery of cells to the brain. In: Eur. J. Cell Biol. 88. Jahrgang, Nr. 6, Juni 2009, S. 315–24, doi:10.1016/j.ejcb.2009.02.001, PMID 19324456.
- ↑ G. Miller: Drug targeting. Breaking down barriers. In: Science 297, 2002, S. 1116–1118. PMID 12183610.
- ↑ M. D. Habgood u. a.: Determinants of passive drug entry into the central nervous system. In: Cell Mol Neurobiol 20, 2000, S. 231–253. PMID 10696512.
- ↑ W. H. Oldendorf: Lipid solubility and drug penetration of the blood-brain barrier. In: Proc Soc Exp Biol Med 147, 1974, S. 813–816. PMID 4445171.
- ↑ W. H. Oldendorf u. a.: Blood-brain barrier: penetration of morphine, codeine, heroin, and methadone after carotid injection. In: Science 178, 1972, S. 984–986. PMID 5084666.
- ↑ D. Patel u. a.: Peptide targeting and delivery across the blood-brain barrier utilizing synthetic triglyceride esters: design, synthesis, and bioactivity. In: Bioconjug Chem 8, 1997, S. 434–441. PMID 9177851.
- ↑ E. Mutschler u. a.: Mutschler Arzneimittelwirkungen. Wissenschaftliche Verlagsgesellschaft, Stuttgart, 2008, ISBN 3-8047-1952-X.
- ↑ Y. Takada u. a.: Rapid high-affinity transport of a chemotherapeutic amino acid across the blood-brain barrier. In: Cancer Res 52, 1992, S. 2191–2196. PMID 1559223.
- ↑ D. M. Killian u. a.: Modulating blood-brain barrier interactions of amino acid-based anticancer agents. In: Drug Deliv 7, 2000, S. 21–25. PMID 10895416.
- ↑ Y. Takada u. a.: Affinity of antineoplastic amino-acid drugs for the large neutral amino acid transporter of the blood–brain barrier. In: Cancer Chemother Pharmacol 30, 1991, S. 89–94. PMID 1760863.
- ↑ A. Bootz: Entwicklung, Charakterisierung und Testung von Nanopartikeln zur Überwindung der Blut-Hirn-Schranke auf Basis von Poly(butylcyanoacrylat). Dissertation, Johann Wolfgang Goethe-Universität, 2006.
- ↑ J. Temsamani u. a.: Vector-mediated drug delivery to the brain. In: Expert Opin Biol Ther 1, 2001, S. 773–782. doi:10.1517/14712598.1.5.773. PMID 11728213 (Review).
- ↑ R. L. Roberts u. a.: Receptor-mediated endocytosis of transferrin at the blood-brain barrier. In: J Cell Sci 104, 1993, S. 521–532. PMID 8505377.
- ↑ B. Dehouck u. a.: Upregulation of the low density lipoprotein receptor at the blood-brain barrier: intercommunications between brain capillary endothelial cells and astrocytes. In: J Cell Biol 126, 1994, S. 465–473. PMID 8034745.
- ↑ K. R. Duffy u. a.: Human blood-brain barrier insulin-like growth factor receptor. In: Metabolism 37, 1988, S. 136–140. PMID 2963191.
- ↑ U. Bickel u. a.: Pharmacologic effects in vivo in brain by vector-mediated peptide drug delivery. In: PNAS 90, 1993, S. 2618–2622. PMID 8385339.
- ↑ T. Moos und E. H. Morgan: Restricted transport of anti-transferrin receptor antibody (OX26) through the blood–brain barrier in the rat. In: Journal of Neurochemistry 79, 2001, S. 119–129. PMID 11595764.
- ↑ W. M. Pardridge u. a.: Human insulin receptor monoclonal antibody undergoes high affinity binding to human brain capillaries in vitro and rapid transcytosis through the blood– brain barrier in vivo in the primate. In: Pharm Res 12, 1995, S. 807–816. PMID 7667183.
- ↑ W. M. Pardridge: Non-invasive drug delivery to the human brain using endogenous blood–brain barrier transport system. In: Pharmacol Sci Technol Today 2, 1999, S. 49–59. PMID 10234207
- ↑ W. M. Pardridge u. a.: Transport of human recombinant brain-derived neurotrophic factor (BDNF) through the rat blood–brain barrier in vivo using vector-mediated peptide drug delivery. In: Pharm Res 11, 1994, S. 738–746. PMID 8058646.
- ↑ a b c J. M. Scherrmann: Drug delivery to brain via the blood-brain barrier. In: Vascul Pharmacol 38, 2002, S. 349–354. PMID 12529929 (Review).
- ↑ D. Karkan u. a.: A unique carrier for delivery of therapeutic compounds beyond the blood-brain barrier. In: PLoS ONE 3, 2008, e2469. PMID 18575595.
- ↑ C. Rousselle u. a.: New advances in the transport of doxorubicin through the blood-brain barrier by a peptide vector-mediated strategy. In: Mol Pharmacol 57, 2000, S. 679–686. PMID 10727512.
- ↑ C. Rousselle u. a.: Enhanced delivery of doxorubicin into the brain via a peptide vector-mediated strategy: saturation kinetics and specificity. In: J Pharmacol Exp Ther 296, 2001, S. 124–131. PMID 11123372.
- ↑ B. Christiaens u. a.: Tryptophan fluorescence study of the interaction of penetratin peptides with model membranes In: FEBS Journal 269, 2002, S. 2918–2926. PMID 12071955.
- ↑ S. R. Schwarze u. a.: In vivo protein transduction: delivery of a biologically active protein into the mouse. In: Science 285, 1999, S. 1569–1572. PMID 10477521.
- ↑ M. Pooga u. a.: Cell penetration by transportan. In: FASEB 12, 1998, S. 67–77. PMID 9438412.
- ↑ M. J. Haney, Y. Zhao, E. B. Harrison, V. Mahajan, S. Ahmed, Z. He, P. Suresh, S. D. Hingtgen, N. L. Klyachko, R. L. Mosley, H. E. Gendelman, A. V. Kabanov, E. V. Batrakova: Specific transfection of inflamed brain by macrophages: a new therapeutic strategy for neurodegenerative diseases. In: PloS one. Band 8, Nummer 4, 2013, S. e61852, doi:10.1371/journal.pone.0061852. PMID 23620794. PMC 3631190 (freier Volltext).
- ↑ E. V. Batrakova, H. E. Gendelman, A. V. Kabanov: Cell-mediated drug delivery. In: Expert opinion on drug delivery. Band 8, Nummer 4, April 2011, S. 415–433, doi:10.1517/17425247.2011.559457. PMID 21348773. PMC 3062753 (freier Volltext).
- ↑ Y. Zhao, M. J. Haney, V. Mahajan, B. C. Reiner, A. Dunaevsky, R. L. Mosley, A. V. Kabanov, H. E. Gendelman, E. V. Batrakova: Active Targeted Macrophage-mediated Delivery of Catalase to Affected Brain Regions in Models of Parkinson’s Disease. In: Journal of nanomedicine & nanotechnology. S4September 2011, S. , doi:10.4172/2157-7439.S4-003. PMID 22288024. PMC 3267477 (freier Volltext).
- ↑ a b F. Hervé u. a.: CNS delivery via adsorptive transcytosis. In: AAPS J 10, 2008, S. 455–472. PMID 18726697 (Review).
- ↑ M. W. Smith und M. Gumbleton: Endocytosis at the blood-brain barrier: from basic understanding to drug delivery strategies. In: J Drug Target 14, 2006, S. 191–214. PMID 16777679 (Review).
- ↑ I. Tamai u. a.: Structure-internalization relationship for adsorptive-mediated endocytosis of basic peptides at the blood– brain barrier. In: J Pharmacol Exp Ther 280, 1997, S. 410–415. PMID 8996222.
- ↑ T. Terasaki u. a.: In vivo transport of a dynorphin-like analgesic peptide, E 2078, through the blood-brain barrier: an application of microdialysis. In: J Pharmacol Exp Ther 251, 1991, S. 815–820. PMID 1681528.
- ↑ S. Nobmann: Isolierte Gehirn-Kapillaren als in vitro-Modell der Blut-Hirn Schranke. Dissertation, Ruprecht-Karls-Universität Heidelberg, 2001.
- ↑ H. H. Szeto: Development of mitochondria-targeted aromatic-cationic peptides for neurodegenerative diseases. In: Ann N Y Acad Sci 1147, 2008, S. 112–121. PMID 19076436.
- ↑ J. F. Poduslo u. a.: Putrescine-modified nerve growth factor: bioactivity, plasma pharmacokinetics, blood– brain/nerve barrier permeability, and nervous system biodistribution. In: J Neurochem 71, 1998, S. 1651–1660. PMID 9751199.
- ↑ T. M. Wengenack u. a.: Putrescine-modified catalase with preserved enzymatic activity exhibits increased permeability at the blood-nerve and blood-brain barriers. In: Brain Res 767, 1997, S. 128–135. PMID 9365024.
- ↑ Vinogradov SV, Bronich TK, Kabanov AV: Nanosized cationic hydrogels for drug delivery: preparation, properties and interactions with cells. In: Adv. Drug Deliv. Rev. 54. Jahrgang, Nr. 1, Januar 2002, S. 135–47, PMID 11755709 (elsevier.com).
- ↑ J. Kreuter: Influence of the surface properties on nanoparticle-mediated transport of drugs to the brain. In: J Nanosci Nanotechnol 4, 2004, S. 484–488. PMID 15503433 (Review).
- ↑ P. Blasi u. a.: Solid lipid nanoparticles for targeted brain drug delivery. In: Adv Drug Deliv Rev 59, 2007, S. 454–477. PMID 17570559 (Review).
- ↑ J. C. Oliver: Drug transport to brain with targeted nanoparticles. In: NeuroRx 2, 2005, S. 108–119. PMID 15717062 (Review).
- ↑ P. Calvo u. a.: Long-circulating PEGylated polycyanoacrylate nanoparticles as new drug carrier for brain delivery. In: Pharm Res 18, 2001, S. 1157–1166. PMID 11587488
- ↑ J. Kreuter: Nanoparticulate systems for brain delivery of drugs. In: Adv Drug Deliv Rev 47, 2001, S. 65–81. PMID 11251246 (Review).
- ↑ J. Kreuter und S. Gelperina: Use of nanoparticles for cerebral cancer. In: Tumori 94, 2008, S. 271–277. PMID 18564616.
- ↑ A. E. Gulyaev u. a.: Significant transport of doxorubicin into the brain with polysorbate 80-coated nanoparticles. In: Pharm Res 16, 1999, S. 1564–1569. PMID 10554098.
- ↑ A. J. Sawyer u. a.: New methods for direct delivery of chemotherapy for treating brain tumors. In: Yale J Biol Med 79, 2006, S. 141–152. PMID 17940624 (Review).
- ↑ E. Garcia-Garcia u. a.: Colloidal carriers and blood-brain barrier (BBB) translocation: a way to deliver drugs to the brain? In: Int J Pharm 298, 2005, S. 274–292. PMID 15896933 (Review).
- ↑ S. B. Tiwari und M. M. Amiji: A review of nanocarrier-based CNS delivery systems. In: Curr Drug Deliv 3, 2006, S. 219–232. PMID 16611008 (Review).
- ↑ I.P. Kaur u. a.: Potential of solid lipid nanoparticles in brain targeting. In: J Control Release 127, 2008, S. 97–109. PMID 18313785 (Review).
- ↑ J. L. Gilmore u. a.: Novel nanomaterials for clinical neuroscience. In: J Neuroimmune Pharmacol 3, 2008, S. 83–94. PMID 18210200 (Review).
- ↑ W. H. De Jong und P. J. Borm: Drug delivery and nanoparticles:applications and hazards. In: Int J Nanomedicine 3, 2008, S. 133–149. PMID 18686775 (Review).
- ↑ R. D. Broadwell u. a.: Morphologic effect of dimethyl sulfoxide on the blood-brain barrier. In: Science 217, 1982, S. 164–166. PMID 7089551.
- ↑ a b c J. P. Hanig u. a.: Ethanol enhancement of blood-brain barrier permeability to catecholamines in chicks. In: Eur J Pharmacol 18, 1972, S. 79–82. PMID 5031276.
- ↑ B Erdlenbruch u. a.: Transient and controllable opening of the blood–brain barrier to cytostatic and antibiotic agents by alkylglycerols in rats. In: Exp. Brain Res. 135, 2000, S. 417–422. PMID 11146820.
- ↑ a b B. Erdlenbruch u. a.: Alkylglycerol opening of the blood-brain barrier to small and large fluorescence markers in normal and C6 glioma-bearing rats and isolated rat brain capillaries. In: British Journal of Pharmacology 140, 2003, S. 1201–1210. PMID 14597599.
- ↑ H. J. Lee u. a.: Blood-brain barrier disruption following the internal carotid arterial perfusion of alkyl glycerols. In: Journal of drug targeting 10, 2002, S. 463–467. PMID 12575736.
- ↑ B. Erdlenbruch u. a.: Intracarotid administration of short-chain alkylglycerols for increased delivery of methotrexate to the rat brain. In: British journal of pharmacology 139, 2003, S. 685–694. PMID 12812991.
- ↑ B. Erdlenbruch u. a.: Blood-brain barrier opening with alkylglycerols: Biodistribution of 1-O-pentylglycerol after intravenous and intracarotid administration in rats. In: J Drug Target 13, 2005, S. 143–150. PMID 16036302.
- ↑ A. Saija u. a.: Changes in the permeability of the blood-brain barrier following sodium dodecyl sulphate administration in the rat. In: Exp Brain Res 115, 1997, S. 546–551. PMID 9262210.
- ↑ M. D. Ellison u. a.: Blood-brain barrier dysfunction in cats following recombinant interleukin-2 infusion. In: Cancer Res 47, 1987, S. 5765–5770. PMID 3499219.
- ↑ M. N. Azmin u. a.: The distribution and elimination of methotrexate in mouse blood and brain after concurrent administration of polysorbate 80. In: Cancer Chemother Pharmacol 14, 1985, S. 238–242. PMID 3995684.
- ↑ T. Sakane u. a.: The effect of polysorbate 80 on brain uptake and analgesic effect of D-kyotorphin. In: Int J Pharm 57, 1989, S. 77–83.
- ↑ a b c Y. Su und P. J. Sinko: Drug delivery across the blood-brain barrier: why is it difficult? how to measure and improve it? In: Expert Opin Drug Deliv 3, 2006, S. 419–435. PMID 16640501 (Review).
- ↑ A. H. Schinkel u. a.: Disruption of the mouse mdr1a P-glycoprotein gene leads to a deficiency in the blood-brain barrier and to increased sensitivity to drugs. In: Cell 77, 1994, S. 491–502. PMID 7910522.
- ↑ P. Jolliet-Riant und J. P. Tillement: Drug transfer across the blood-brain barrier and improvement of brain delivery. In: Fundam Clin Pharmacol 13, 1999, S. 16–26. PMID 10027084 (Review).
- ↑ R. B. Kim u. a.: The drug transporter P-glycoprotein limits oral absorption and brain entry of HIV-1 protease inhibitors. In: J Clin Investig 101, 1998, S. 289–294. PMID 9435299.
- ↑ E. M. Kemper u. a.: Increased penetration of paclitaxel into the brain by inhibition of P-glycoprotein. In: Clin Cancer Res 9, 2003, S. 2849–2855. PMID 12855665.
- ↑ a b E. M. Kemper u. a.: The influence of the P-glycoprotein inhibitor zosuquidar trihydrochloride (LY335979) on the brain penetration of paclitaxel in mice. In: Cancer Chemother Pharmacol 53, 2004, S. 173–178. PMID 14605863.
- ↑ a b E. M. Kemper u. a.: Improved penetration of docetaxel into the brain by co-administration of inhibitors of P-glycoprotein. In: European Journal of Cancer 40, 2004, S. 1269–1274. PMID 15110893.
- ↑ a b U. Mayer u. a.: Full blockade of intestinal P-glycoprotein and extensive inhibition of blood-brain barrier P-glycoprotein by oral treatment of mice with PSC833. In: J Clin Investig 100, 1997, S. 2430–2436. PMID 9366556.
- ↑ a b N. H. Hendrikse u. a.: Complete in vivo reversal of P-glycoprotein pump function in the blood-brain barrier visualized with positron emission tomography. In: Br J Pharmacol 124, 1998, S. 1413–1418. PMID 9723952.
- ↑ H. Kusuhara u. a.: P-Glycoprotein mediates the efflux of quinidine across the blood-brain barrier. In: J Pharmacol Exp Ther 283, 1997, S. 574–580. PMID 9353372.
- ↑ P. Hsiao u. a.: Verapamil P-glycoprotein transport across the rat blood-brain barrier: cyclosporine, a concentration inhibition analysis, and comparison with human data. In: J Pharmacol Exp Ther. 317, 2006, S. 704–710. PMID 16415090.
- ↑ N. Drion u. a.: Role of P-glycoprotein in the blood-brain transport of colchicine and vinblastine. In: J Neurochem 67, 1996, S. 1688–1693. PMID 8858954.
- ↑ I. Sauer: Apolipoprotein E abgeleitete Peptide als Vektoren zur Überwindung der Blut-Hirn-Schranke. Dissertation FU Berlin, 2004.
- ↑ L. He, C. Zhao, M. Yan, L. Y. Zhang, Y. Z. Xia: Inhibition of P-glycoprotein function by procyanidine on blood-brain barrier. In: Phytother Res 23, 2009, S. 933–937 PMID 19172664.
- ↑ S. F. Zhou: Structure, function and regulation of P-glycoprotein and its clinical relevance in drug disposition. In: Xenobiotica 38, 2008, S. 802–832. PMID 18668431 (Review).
- ↑ Y. J. Lee u. a.: Investigation of efflux transport of dehydroepiandrosterone sulfate and mitoxantrone at the mouse blood-brain barrier: a minor role of breast cancer resistance protein. In: J Pharmacol Exp Ther 312, 2005, S. 44–52. PMID 15448171.
- ↑ H. Yuan u. a.: Strategies to overcome or circumvent P-glycoprotein mediated multidrug resistance. In: Curr Med Chem 15, 2008, S. 470–476. PMID 18289002 (Review).
- ↑ H. M. Coley: Mechanisms and strategies to overcome chemotherapy resistance in metastatic breast cancer. In: Cancer Treat Rev 34, 2008, S. 378–390. PMID 18367336 (Review).
- ↑ S. Nobili u. a.: Pharmacological strategies for overcoming multidrug resistance. In: Curr Drug Targets 7, 2006, S. 861–879. PMID 16842217 (Review).
- ↑ a b N. N. Salama u. a.: Tight junction modulation and its relationship to drug delivery. In: Adv Drug Deliv 58, 2006, S. 15–28. PMID 16517003.
- ↑ N. S. Ningaraj Drug delivery to brain tumours: challenges and progress. In: Expert Opin Drug Deliv 3, 2006, S. 499–509. PMID 16822225.
- ↑ M. Kondoh und K. Yagi: Tight junction modulators: promising candidates for drug delivery. In: Curr Med Chem 14, 2007, S. 2482–2488. PMID 17979701 (Review).
- ↑ P. H. Johnson u. a.: Discovery of tight junction modulators: significance for drug development and delivery. In: Drug Discov Today 13, 2008, S. 261–267.PMID 18342803 (Review).
- ↑ A. Fasano u. a.: Zonula occludens toxin modulates tight junctions through protein kinase C-dependent actin reorganization, in vitro. In: J Clin Invest 96, 1995, S. 710–720. PMID 7635964.
- ↑ M. A. Deli: Potential use of tight junction modulators to reversibly open membranous barriers and improve drug delivery. In: Biochim. Biophys. Acta 1788, 2009, S. 892–910 PMID 18983815 (Review).
- ↑ C.S. Karyekar u. a.: Zonula occludens toxin increases the permeability of molecular weight markers and chemotherapeutic agents across the bovine brain microvessel endothelial cells. In: J Pharm Sci 92, 2003, S. 414–423. PMID 12532391.
- ↑ K.-H. Song u. a.: Effect of the six-mer synthetic peptide (AT1002) fragment of zonula occludens toxin on the intestinal absorption of cyclosporin A. In: Int J Pharm 351, 2008, S. 8–14. PMID 17954018.
- ↑ V. Wong und B. Gumbiner: A synthetic peptide corresponding to the extracellular domain of occludin perturbs the tight junction permeability barrier. In: J Cell Biol 136, 1997, S. 399–409. PMID 9015310.
- ↑ S. Zausinger: Bradykinin receptor antagonists in cerebral ischemia and trauma. In: IDrugs 6, 2003, S. 970–975. PMID 14534854 (Review).
- ↑ a b N. Hettenbach: Einfluss chronischer elektromagnetischer Befeldung mit Mobilfunkstrahlen (GSM und UMTS) auf die Integrität der Blut-Hirn-Schranke von Ratten. Dissertation, Ludwig-Maximilians-Universität München, 2008.
- ↑ S. I. Rapoport u. a.: Testing of a hypothesis for osmotic opening of the blood-brain barrier. In: Am J Physiol 223, 1972, S. 323–331. PMID 5046750.
- ↑ a b S. I. Rapoport u. a.: Quantitative aspects of reversible osmotic opening of the blood-brain barrier. In: Am. J. Physiol. 238, 1980, S. 421–431. PMID 7377381.
- ↑ K. Dorovini-Zis: Hyperosmotic arabinose solutions open the tight junctions between brain capillary endothelial cells in tissue culture. In: Brain Res 302, 1984, S. 383–386. PMID 6733518.
- ↑ S. I. Rapoport: Effect of concentrated solutions on the blood-brain barrier. In: Am J Physiol 219, 1970, S. 270–274. PMID 5424853.
- ↑ S. I. Rapaport u. a.: Reversible osmotic opening of the blood-brain barrier. In: Science 173, 1971, S. 1026–1028. PMID 5098961.
- ↑ E. A. Neuwelt u. a.: Use of enhanced computerized tomography to evaluate osmotic blood-brain barrier disruption. In: Neurosurgery 6, 1980, S. 49–56. PMID 6153461.
- ↑ Y. Z. Zilyan u. a.: Blood-brain barrier permeability to sucrose and dextran after osmotic opening. In: Am J Physiol 247, 1984, S. R634–R638. PMID 6208789.
- ↑ P. J. Robinson und S. I. Rapoport: Model for drug uptake by brain tumors: Effects of osmotic treatment and of diffusion in brain. In: J Cereb Blood Flow Metab 10, 1990, S. 153–161. PMID 2303532.
- ↑ a b P. J. Robinson und S. I. Rapoport: Size selectivity of blood-brain barrier permeability at various times after osmotic opening. In: Am J Physiol 253, 1987, S. R459–R466. PMID 2443025.
- ↑ S. I. Rapoport: Osmotic opening of the blood-brain barrier: principles, mechanism, and therapeutic applications. In: Cell Mol Neurobiol 20, 2000, S. 217–230. PMID 10696511.
- ↑ S. I. Rapoport: Modulation of the blood-brain barrier permeability. In: Journal of Drug Targeting 3, 1996, S. 417–425. PMID 8863135.
- ↑ S. I. Rapoport: Advances in osmotic opening of the blood-brain barrier to enhance CNS chemotherapy. In: Expert Opin Investig Drugs 10, 2001, S. 1809–1818. PMID 11772287 (Review).
- ↑ K. Jahnke u. a.: Intraarterial chemotherapy and osmotic blood-brain barrier disruption for patients with embryonal and germ cell tumors of the central nervous system. In: Cancer 112, 2008, S. 581–588. PMID 18072268.
- ↑ L. Bakay u. a.: Ultrasonically produced changes in the blood–brain barrier. In: Arch Neurol Psychiatry 76, 1956, S. 457–467. PMID 13371961.
- ↑ H. T. Ballantine u. a.: Progress and problems in the neurological applications of focused ultrasound. In: J Neurosurg 17, 1960, S. 858–876. PMID 13686380.
- ↑ K. Hynynen u. a: Noninvasive MR imaging-guided focal opening of the blood–brain barrier in rabbits. In: Radiology 220, 2001, S. 640–646. PMID 11526261.
- ↑ S. Meairs und A. Alonso: Ultrasound, microbubbles and the blood-brain barrier. In: Prog Biophys Mol Biol 93, 2007, S. 354–362. PMID 16959303 (Review).
- ↑ H. L. Liu u. a.: Magnetic resonance imaging enhanced by superparamagnetic iron oxide particles: usefulness for distinguishing between focused ultrasound-induced blood-brain barrier disruption and brain hemorrhage. In: J Magn Reson Imaging 29, 2009, S. 31–38. PMID 19097103.
- ↑ N. McDannold u. a.: Effects of acoustic parameters and ultrasound contrast agent dose on focused-ultrasound induced blood-brain barrier disruption. In: Ultrasound Med Biol 34, 2008, S. 930–937. PMID 18294757.
- ↑ G. T. Clement und K. Hynynen: A non-invasive method for focusing ultrasound through the human skull. In: Phys Med Biol 47, 2002, S. 1219–1236. PMID 12030552.
- ↑ a b K. Hynynen: Ultrasound for drug and gene delivery to the brain. In: Advanced Drug Delivery Reviews 60, 2008, S. 1209–1217. PMID 18486271 (Review).
- ↑ a b W. L. Nyborg: Biological effects of ultrasound: development of safety guidelines. Part II: general review. In: Ultrasound Med Biol 27, 2001, S. 301–333. PMID 11369117.
- ↑ L. Krizanac-Bengez u. a.: The cerebral vasculature as a therapeutic target for neurological disorders and the role of shear stress in vascular homeostatis and pathophysiology. In: Neurol Res 26, 2004, S. 846–853. PMID 15727268 (Review).
- ↑ N. Sheikov u. a.: Effect of focused ultrasound applied with an ultrasound contrast agent on the tight junctional integrity of the brain microvascular endothelium. In: Ultrasound Med Biol 34, 2008, S. 1093–1104. PMID 18378064.
- ↑ L. H. Treat u. a.: Targeted delivery of doxorubicin to the rat brain at therapeutic levels using MRI-guided focused ultrasound. In: Int J Cancer 121, 2007, S. 901–907. PMID 17437269.
- ↑ M. Kinoshita u. a.: Noninvasive localized delivery of Herceptin to the mouse brain by MRI-guided focused ultrasound-induced blood-brain barrier disruption. In: PNAS 103, 2006, S. 11719–11723. PMID 16868082.
- ↑ M. Kinoshita u. a.: Targeted delivery of antibodies through the blood-brain barrier by MRI-guided focused ultrasound. In: Biochem Biophys Res Commun 340, 2006, S. 1085–1090. PMID 16403441.
- ↑ S. B. Raymond u. a.: Ultrasound Enhanced Delivery of Molecular Imaging and Therapeutic Agents in Alzheimer’s Disease Mouse Models. In: PLoS ONE 3, 2008, e2175. PMID 18478109.
- ↑ N. Sheikov u. a.: Brain arterioles show more active vesicular transport of blood-borne tracer molecules than capillaries and venules after focused ultrasound-evoked opening of the blood-brain barrier. In: Ultrasound Med Biol 32, 2006, S. 1399–1409. PMID 16965980.
- ↑ G. J. Jungehulsing u. a.: Diagnostic transcranial ultrasound perfusion-imaging at 2.5 MHz does not affect the blood-brain barrier. In: Ultrasound Med Biol 34, 2008, S. 147–150. PMID 17854981.