Naturstoff (englisch natural product) bezeichnet in der Chemie eine Verbindung, die von Organismen gebildet wird, um biologische Funktionen zu erfüllen; ein modernes Synonym für Naturstoff im Sinn der Chemie ist Biomolekül. Dabei werden nur Reinsubstanzen oder definierte Stoffgemische als solche verstanden. Der Gesetzgeber definiert Biostoffe mittels der Biostoffverordnung.
Im allgemeinen Sprachgebrauch ist Naturstoff ein weiter gefasster Begriff: Alle Stoffe, die der Mensch nicht künstlich hergestellt hat. Dieser Artikel bezieht sich nur auf den zuvor definierten speziellen Begriff im Sinn der Chemie. Danach zählen nicht zu den Naturstoffen
- von Lebewesen gebildete komplexe, nicht reine Stoffe, Naturprodukte wie Federn, Holz oder Baumwolle, die zwar aus Naturstoffen aufgebaut, jedoch uneinheitliche Stoffgemische sind
- alle natürlich vorkommenden anorganischen Stoffe wie Minerale oder Gesteine
Obwohl es sich bei einem Naturstoff um eine definierte Verbindung handelt, kommt er in Organismen in vielen ineinander überführbaren Modifikationen vor. Durch die Modifikationen wird die biologische Funktion kontrolliert bzw. gesteuert. Nach der Extraktion und Reinigung erhält man meist die stabile Grundstruktur, die man den Naturstoffklassen zuordnen kann.
Naturstoffe werden in allen lebenden Organismen aufgebaut oder ineinander umgewandelt. Ihre Synthese ist für den Organismus mit einem Energieaufwand verbunden. Ihre Aufgaben sind je nach Stoffklasse vielfältig und reichen vom einfachen Stoffwechsel oder der Energiegewinnung über Zellbestandteile und Baustoffe des Organismus bis zu komplexen Steueraufgaben. In Bezug auf ihre Funktionen kann man zwischen primären und sekundären Naturstoffen unterscheiden. Zu den primären Naturstoffen werden alle Verbindungen gezählt, die für den Organismus zur Lebenserhaltung und zum Wachstum benötigt werden. Dazu zählen vor allem die Fette und die Biopolymere der Kohlenhydrate und Proteine. Die sekundären Naturstoffe werden aus häufig noch unbekannten Gründen gebildet und unterteilen sich in die großen Stoffklassen der Terpene, der Aromaten und der Alkaloide.
Die Chemie der Naturstoffe ist eine interdisziplinäre Wissenschaft, die mit Methoden der organischen und analytischen Chemie Fragestellungen der Biologie, Biochemie, Physiologie und der Pharmazie aufklären kann. Die Naturstoffchemie hat eine große Bedeutung in der Pharmakologie, bei der Entwicklung neuer Wirkstoffe und generell bei der Methodenentwicklung in diesen Disziplinen.
Geschichte
Bearbeiten
Der ursprüngliche Begriff Naturstoff wurde durch die historische Definition der organischen Chemie bestimmt; sie umfasste die Gesamtheit der Stoffe, die dem Aufbau von Tieren und Pflanzen dienen. Noch Jöns Jakob Berzelius nahm 1827 auf Grund des Wissenstandes und des komplizierten chemischen Aufbaus der Naturstoffe an, dass es für ihre Erzeugung eine Lebenskraft (vis vitalis) geben müsse. Die Unterscheidung zwischen selbstorganisierten und fremdorganisierten Stoffen wurde durch Friedrich Wöhler (1828) revidiert, der mit der Harnstoffsynthese bewiesen hatte, dass aus der als anorganisch definierten Verbindung Ammoniumcyanat die als organisch definierte Verbindung Harnstoff hergestellt werden kann.

Zur organischen Chemie gehört in der heutigen Definition praktisch die Gesamtheit der Kohlenstoffverbindungen. Die Naturstoffchemie entwickelte sich im Laufe der Zeit zu deren Teilgebiet und beschäftigt sich mit der Isolierung, Strukturaufklärung, Synthese bzw. Biosynthese und den Eigenschaften von Verbindungen, die in Organismen (wie Tieren, Pflanzen und Mikroorganismen) vorkommen.
Zunächst befasste sich die Naturstoffchemie nur mit Inhaltsstoffen pflanzlicher Herkunft, da sie sehr von der Pharmakognosie (Drogenkunde) geprägt war. Aus Pflanzenextrakten wurden Verbindungen (zunächst meist Alkaloide) isoliert und man versuchte, deren Struktur aufzuklären.
Justus Liebig weitete dann den Naturstoffbegriff zur Mitte des 19. Jahrhunderts auch auf Verbindungen tierischer Herkunft aus. Emil Fischer wurde gegen Ende des 19. Jahrhunderts ein Pionier bei der Strukturaufklärung und Synthese der Kohlenhydrate und Proteine.
Bis Ende der 1930er Jahre wurden die wichtigsten Naturstoffklassen gefunden, untersucht und deren Struktur aufgeklärt. Wichtige Meilensteine sind hier:
- Terpene durch Otto Wallach aus ätherischen Ölen
- Steroide durch Adolf Windaus und Heinrich Otto Wieland
- Carotinoide durch Paul Karrer
- Porphinfarbstoffe durch Richard Willstätter und Hans Fischer
- Vitamine unter anderem durch Paul Karrer, Adolf Windaus, Robert R. Williams, Richard Kuhn und Albert von Szent-Györgyi Nagyrápolt
- Hormone durch Adolf Butenandt und Edward Calvin Kendall
Mit der Entdeckung des Penicillins im Jahr 1940 durch Alexander Fleming, Ernst Boris Chain und Howard Walter Florey wurden auch Mikroorganismen als lohnende Quelle für Naturstoffe erkannt.
Die Naturstoffchemie wurde nach dem Ende des Zweiten Weltkriegs durch die Entwicklung von neuen und sehr leistungsfähigen analytischen und physikalischen Methoden vorangetrieben. Sowohl die Massenspektrometrie, Röntgenstrukturanalyse und später auch die NMR-Spektroskopie ermöglichten bis dahin ungeahnte Möglichkeiten der Strukturaufklärung ohne Derivatisierung des Naturstoffes und bei geringen Mengen des Analyten. Die erst jetzt langsam etablierten Methoden der Chromatographie und Elektrophorese erlaubten eine Trennung der Stoffgemische in bisher nicht erreichbarer Geschwindigkeit und Reinheit.[1][2]
Definition im Chemikalienrecht
BearbeitenDer Begriff Naturstoff wird juristisch definiert als „natürlich vorkommender Stoff als solcher, unverarbeitet oder lediglich manuell, mechanisch oder durch Gravitationskraft, durch Auflösung in Wasser, durch Flotation, durch Extraktion mit Wasser, durch Dampfdestillation oder durch Erhitzung zum Wasserentzug verarbeitet oder durch beliebige Mittel aus der Luft entnommen“.[3][4]
Nach Anhang V Absätze 7 und 8 der REACH-Verordnung (EG 1907/2006) sind Naturstoffe von einer Registrierungspflicht ausgenommen, wenn sie nicht chemisch verändert wurden. Dies gilt nicht, wenn sie nach den Kriterien der CLP-Verordnung (EG Nr. 1272/2008) als gefährlich eingestuft werden, die Kriterien für PBT- bzw. vPvB-Stoffe erfüllen oder ebenso besorgniserregend sind.[4]
Definition aus der Biostoffverordnung
BearbeitenBiologische Arbeitsstoffe sind rechtlich nach der BioStoffV im Wesentlichen Mikroorganismen, Proteinstrukturen, welche Erkrankungen hervorrufen können und Zellkulturen.[5]
„Die Biostoffverordnung setzt neben der europäischen Richtlinie 2000/54/EG (Schutz der Arbeitnehmer gegen Gefährdung durch biologische Arbeitsstoffe bei der Arbeit) auch die europäische Richtlinie 2010/32/EU (Vermeidung von Verletzungen durch scharfe/spitze Instrumente im Krankenhaus- und Gesundheitssektor) in deutsches Arbeitsschutzrecht um.
Die Biostoffverordnung regelt berufsbedingte Tätigkeiten mit biologischen Arbeitsstoffen, d. h. im weitesten Sinne mit Mikroorganismen/Krankheitserregern. Sie enthält Regelungen zum Schutz der Beschäftigten bei diesen Tätigkeiten, d. h. zum Schutz vor Infektionen sowie vor sensibilisierenden, toxischen oder anderen die Gesundheit schädigenden Wirkungen. Die Biostoffverordnung teilt biologische Arbeitsstoffe in vier Risikogruppen ein. Auf dieser Basis erfolgt die Gefährdungsbeurteilung und die Festlegung der erforderlichen Maßnahmen.“
Bedeutung der Naturstoffchemie
BearbeitenPharmakologie
BearbeitenNaturstoffe sind spätestens seit der Entdeckung des Penicillins durch Alexander Fleming[6] zu einer wichtigen Quelle für Leitstrukturen pharmazeutischer Wirkstoffe geworden. Man findet bei Naturstoffen pharmakologische Wirkungen als Antibiotika, Immunsuppressiva, Enzym-Inhibitoren, Rezeptor-Antagonisten und -Agonisten, Toxine, antitumorale und antivirale Wirkstoffe.
Eine ganze Reihe von Wirkstoffen leiten sich heute von Naturstoffen ab. Dazu gehören neben β-Lactam-Antibiotika auch Chemotherapeutika wie Paclitaxel aus der Pazifischen Eibe (Taxus brevifolia)[7] oder Epothilon aus dem Myxobakterium Sorangium cellulosum.[8]
Nucleoside wurden so modifiziert, dass sie als Virustatika Verwendung finden, wie etwa der HIV-Wirkstoff Zidovudin.[9]
Die jahrhundertelange Erfahrung aus der Volksmedizin hat die Aufmerksamkeit auf viele Pflanzen und damit auf ihre Inhaltsstoffe als Leitstrukturen gelenkt. So sind beispielsweise schon seit langem Ginseng (Panax ginseng),[10][11] Ginkgo (Ginkgo biloba)[12] oder der Niembaum (Azadirachta indica)[13] Objekt intensiver Untersuchungen.
-
 Pazifische Eibe (Taxus brevifolia)
Pazifische Eibe (Taxus brevifolia) -
 Wurzeln des Ginsengs (Panax ginseng)
Wurzeln des Ginsengs (Panax ginseng) -
 Blätter des Ginkgos (Ginkgo biloba)
Blätter des Ginkgos (Ginkgo biloba) -
 Niembaum (Azadirachta indica)
Niembaum (Azadirachta indica)




Die so erhaltenen Leitstrukturen dienen als Grundlage für die pharmakologische Optimierung des Wirkstoffes. Dabei werden Struktur-Wirkungs-Beziehungen (QSAR) aufgestellt und versucht, die physikalischen Eigenschaften wie die Löslichkeit in wässrigen Medien zu optimieren. Dazu werden häufig Techniken wie die Parallelsynthese oder die der kombinatorischen Chemie angewandt.[14][15]
Bei Naturstoffen wird pharmakologisch von privilegierten Strukturen gesprochen, da sie unter physiologischen Bedingungen gebildet werden und vorteilhafte pharmakokinetische Eigenschaften zeigen.[16] Die Tatsache, dass etwa die Hälfte der meistverkauften Wirkstoffe Naturstoffe oder deren Derivate sind, zeigt die besondere Bedeutung der Naturstoffe für die Pharmakologie auf.[17]
Biologie
BearbeitenFür die Biologie ist die Aufklärung von physiologischen Zusammenhängen wichtig. Dazu gehören auf dem Gebiet der Naturstoffe die Biosynthese und die biologische Funktion in Organismen, sei es als Enzym, Botenstoff oder als Energielieferant bzw. -speicher. Zu den Botenstoffen gehören u. a. Hormone, Neurotransmitter und Pheromone. Energielieferanten und -speicher sind in der Regel Fette, Eiweiße und Kohlenhydrate. Die Biosynthese von Naturstoffen ist so vielfältig wie ihre Varianz in der Struktur.
Die Aufklärung eines Biosynthesewegs erfordert häufig großen Aufwand. Dabei werden verschiedene Techniken eingesetzt:[18][19]
- Isotopentechnik – Potentielle Vorläufermoleküle werden mit einem seltenen, ggf. auch radioaktiven, aber nicht zu schnell zerfallenden Isotop, wie zum Beispiel dem 14C-Isotop (Halbwertzeit 5736 Jahre), markiert. Nach dem Einschleusen in den Stoffwechsel (z. B. durch Verfüttern oder Einspritzen) wird der Verbleib des Isotops im Zielmolekül beobachtet. Daraus kann auf den Weg der Biosynthese geschlossen werden.
- Enzymatische Techniken – Hier muss der Biosyntheseweg annähernd bekannt sein. Man arbeitet an isolierten Enzymen oder mit Zellkulturen, um Biosynthesewege unter Laborbedingungen und nicht mehr in vivo zu untersuchen.
- Gentechnische Methoden – Beispielsweise wird hier die Biosynthese durch Bakterien mit Hilfe von Genmutationen unterbrochen; ein Intermediat einer Biosynthesesequenz reichert sich dabei an.
Organische Chemie
Bearbeiten
Für die organische Chemie stellt die Naturstoffchemie in vielerlei Hinsicht eine Herausforderung dar. Zum einen auf dem Gebiet der Strukturaufklärung, also der Analytik: Nicht selten arbeiten an der Strukturaufklärung mehrere Arbeitsgruppen über Jahre. Komplexe Strukturen wie z. B. das Azadirachtin bedurften mehrerer Anläufe, bis die korrekte Struktur bewiesen wurde.[20] Auch die Totalsynthese eines Naturstoffes beweist nicht immer die richtige Struktur – wie sich am Patchulialkohol und der Synthese von Büchi gezeigt hat.[21][22]
Generell ist auch die Synthese eines komplexen Naturstoffes eine Herausforderung für die organische Chemie. Um als Pharmakon eine Bedeutung zu erlangen, muss für einen Naturstoff eine Totalsynthese oder Partialsynthese entwickelt werden. Noch wichtiger ist dies im Rahmen der Strukturoptimierung, denn dabei müssen tausende Verbindungen auf der Basis eines Naturstoffes synthetisiert werden, um eine Struktur-Wirkungs-Beziehung aufstellen und die pharmakologischen Eigenschaften optimieren zu können.[23] Naturstoffe sind in der Regel komplexe Verbindungen mit Chiralitätszentren, welche in der gewünschten stereochemischen Konfiguration aufgebaut werden müssen. Die verwendeten Reagenzien müssen kompatibel mit den funktionellen Gruppen im Molekül sein, oder es muss zusätzlich eine entsprechende Schutzgruppenstrategie gewählt werden.
Wie lange und arbeitsintensiv eine Naturstoffsynthese sein kann, zeigen beispielsweise die Totalsynthesen von Vitamin B12 durch Robert B. Woodward und Albert Eschenmoser aus dem Jahr 1973,[24] die Synthese von Palytoxin durch Yoshito Kishi aus dem Jahr 1994[25] oder der Wettlauf um die erste Totalsynthese von Taxol zwischen Robert A. Holton, Kyriacos C. Nicolaou und Samuel J. Danishefsky aus demselben Jahr.[26] Die Synthese von Vitamin B12 hat etwa 20 Jahre Entwicklungsarbeit erfordert. Hierfür mussten jeweils ganz neue Reaktionsschritte entwickelt werden und beim Vitamin B12 mit den Woodward-Hoffmann-Regeln sogar neue theoretische Grundlagen geschaffen werden. Für deren Entwicklung wurde Roald Hoffmann mit dem Nobelpreis gewürdigt.[27] Robert B. Woodward war schon im Jahr 1965 für seine Arbeiten auf dem Gebiet der Naturstoffchemie ausgezeichnet worden.[28][29][30]
Eine weitere Bedeutung von Naturstoffen in der organischen Chemie ist ihre Nutzung als Quelle für Synthesebausteine. Sehr viele Naturstoffe, wie z. B. Zucker oder Aminosäuren, sind chirale Verbindungen und können so als Vorläufermoleküle für chirale Synthesen oder als Reagenzien benutzt werden. Naturstoffe können aber auch einfach eine Quelle für komplexe Ausgangsverbindungen und sogar für industrielle Synthesen darstellen. So stellt beispielsweise die Shikimisäure das Startmaterial für die großtechnische Synthese des Grippewirkstoffs Oseltamivir (Tamiflu) der Firma Roche dar.[31]
Klassifikation nach biologischer Funktion
BearbeitenBei der Klassifikation von Naturstoffen nach biologischer Funktion unterscheidet man zwischen primären Naturstoffen und sekundären Naturstoffen. Die Unterscheidung geht auf den Nobelpreisträger Albrecht Kossel zurück.[32][33]
Diese Einteilung ist heute eher willkürlich und historisch bedingt, wird jedoch immer noch in der Literatur verwendet. Sowohl von der chemischen Struktur als auch von der biologischen Funktion ist diese Gliederung überholt, da ein Naturstoff sowohl eine lebenserhaltende Funktion im Sinne von Kossel haben kann, aber auch klassische Funktionen der sekundären Naturstoffe (Transmittermoleküle, Pheromone, Fraßabwehr usw.) haben kann.
Primäre Naturstoffe
BearbeitenZu den primären Naturstoffen zählen nach der Definition von A. Kossel alle Verbindungen, die im Organismus für den Lebenserhalt und das Wachstum notwendig sind. Es handelt sich hierbei aber um keine streng abgegrenzte Klasse, und die Übergänge zwischen den primären und sekundären Stoffwechselwegen sind fließend.[34]
Primäre Naturstoffe findet man beim Aufbau (Wachstum) von Lebewesen, aber auch während des Abbaus zu kleineren Molekülen, was mit einem Energiegewinn für den Organismus einhergehen kann. Diese Energie kann wiederum zum Aufbau von anderen primären oder sekundären Biomolekülen verwendet werden. Aufbau und Abbau von Naturstoffen sind die Grundlage für den Energie- und Massestoffwechsel in allen Organismen.
Sekundäre Naturstoffe
BearbeitenSekundäre Naturstoffe werden aus vielen Gründen gebildet, sind jedoch nicht essentiell für den Lebenserhalt des Organismus. Sie stellen insbesondere als sekundäre Pflanzenstoffe eine sehr große Vielfalt an chemischen Strukturen und werden im sogenannten Sekundärstoffwechsel gebildet. Dieser schließt sich an den primären Stoffwechsel an und kann daher nicht unabhängig von diesem stattfinden. Der sekundäre Stoffwechsel ist jedoch nicht am Energiestoffwechsel beteiligt und ist weder Bestandteil des anabolen (aufbauenden) noch des katabolen (abbauenden) Stoffwechsels. Sekundäre Naturstoffe werden nur in speziellen Zelltypen gebildet. Die Übergänge von primären Stoffwechselprodukten zu sekundären Stoffwechselprodukten sind fließend. Die biologische Funktion von sekundären Naturstoffen ist sehr vielfältig und auch häufig nicht bekannt.
Klassifikation nach chemischer Struktur
BearbeitenBei der Klassifikation von Naturstoffen nach chemischer Struktur (Stoffklasse) unterscheidet man die Biomoleküle und Biomolekülgruppen nach ihren funktionellen Gruppen, d. h. ihrem strukturellen Aufbau. Bei Naturstoffen findet man sowohl kleine, einfache Moleküle wie z. B. Steroide, Aromatische Verbindungen oder Fettsäuren, aber auch sehr komplexe Biopolymere wie Proteine, DNA und Kohlenhydrate. Auch diese Klassifizierung ist nicht als absolut anzusehen, da komplex zusammengesetzte Stoffe, z. B. ein hochgradig glycosyliertes Protein (Glycoprotein), nicht einer reinen chemischen Stoffklasse zugeordnet werden können.
Aminosäuren, Peptide und Proteine
BearbeitenDie proteinogenen Aminosäuren sind ausschließlich α-Aminosäuren. Es kommen jedoch auch β-Aminosäuren wie β-Alanin, β-Aminobuttersäure oder γ-Aminobuttersäure natürlich vor. Alle natürlich vorkommenden α-Aminosäuren (mit Ausnahme des Glycins) sind chiral. Es handelt sich praktisch ausschließlich um L-Aminosäuren.[35]
Die vollständige Synthese aller 20 biogenen kanonischen Aminosäuren findet man nur in Mikroorganismen und Pflanzen. Tiere – also auch der Mensch – müssen bestimmte Aminosäuren – beim Menschen sind dies Valin, Leucin, Isoleucin, Methionin, Threonin, Lysin, Phenylalanin und Tryptophan,[36] im Kindesalter zusätzlich Tyrosin – als essentielle Aminosäuren mit der Nahrung aufnehmen. Für Fische und Insekten sind zusätzlich auch noch Arginin und Histidin essentiell.
Grundlage für alle Varianten von Aminosäuren bilden die proteinogenen Aminosäuren. Weitere Aminosäuren wie z. B. Ornithin oder Homoserin kommen in Proteinen und als Stoffwechselprodukte vor. Weitere nicht proteinogene Aminosäuren werden durch Hydroxylierung von proteinogenen Aminosäuren gebildet. Dazu gehört z. B. 4-Hydroxyprolin. Des Weiteren findet man Produkte aus N-Methylierungen oder Iodierungen. In Mollusken wurden einige halogenierte Aminosäuren gefunden. Insgesamt wurden bisher über 400 Aminosäuren identifiziert, die nicht in Proteine eingebaut werden. Viele davon werden durch Hydroxylierung oder Methylierung von homologen proteinogenen Aminosäuren gebildet. Sie kommen in Peptidantibiotika oder als Toxine (wie z. B. im Knollenblätterpilz) vor. Seltene Aminosäuren wie Canavanin (Fabaceae), Mimosin (Mimosen-Arten) und 2-Methylen-cyclopropylglycin (Sapindaceae) wirken als Antagonisten der strukturverwandten Aminosäuren Arginin, Phenylalanin und Tyrosin bzw. Leucin und sind daher toxisch.
Peptide und Proteine
BearbeitenSowohl Peptide als auch Proteine sind Ketten von Aminosäuren, welche über eine Amidbindung miteinander verknüpft sind. Man unterscheidet hier Oligopeptide, Peptide und Proteine je nach Anzahl der Aminosäuren und der Molmasse.
| Name | Anzahl der Aminosäuren | Molmasse |
|---|---|---|
| Oligopeptid | 2–10 Aminosäuren | |
| Peptid | >10 bis etwa 80–90 Aminosäuren | |
| Protein | ab etwa 80–90 Aminosäuren | ab 10.000 Da bzw. 10 kDa |


Die Unterteilung zwischen Peptid und Protein beruht darauf, dass Proteine infolge ihrer hohen Molmasse keine Dialysemembranen passieren können. Da die Molekülmassen von Proteinen recht groß sind, benutzt man hier gebräuchlicherweise die Maßeinheit Kilodalton (Einheitenzeichen kDa), welches der normalen gebräuchlichen Masseneinheit für Atome und Moleküle entspricht, jedoch um das Präfix „Kilo“ (und damit um den Faktor 103) erweitert ist.
Aufgrund der vektoriellen Verknüpfung eines acylischen Peptides oder Proteins unterscheidet man zwischen den beiden Enden, dem N-Terminus (dem Ende mit freier oder modifizierter Aminogruppe) und dem C-Terminus (dem Ende mit freier Carboxylatgruppe).
Peptide, welche nur aus Aminosäuren aufgebaut sind, werden als homöomere Peptide bezeichnet. Peptide, die auch Pseudoaminosäuren enthalten, werden als heteromere Peptide bezeichnet. Zu den Pseudoaminosäuren werden z. B. Hydroxycarbonsäuren gerechnet, welche die alternierende Amidstruktur eines Peptides durch eine Esterbindung unterbrechen.
Cyclische Peptide werden auch Peptolide genannt. Je nachdem, ob in einem Peptid die Aminosäuren nur über Amidbindungen untereinander verknüpft sind oder ob noch weitere Bindungen vorhanden sind, wie beispielsweise Disulfid-Brücken, spricht man im ersten Fall von homodeten Peptiden und sonst von herodeten Peptiden. Sehr viele Peptide sind streng linear aufgebaut – aber es gibt auch verzweigte Peptide, die durch Reaktionen an den Seitenketten gebildet werden. Die Ribonukleasen sind ein Beispiel für verzweigte Peptide.[37]
Aufgrund ihres modularen Aufbaus sind Proteine und Peptide sehr variabel in ihren physikalischen Eigenschaften und haben deshalb in Organismen sehr spezielle und sehr unterschiedliche Funktionen. Zu den wichtigen Aufgaben von Proteinen gehören die als Enzym, das heißt, sie katalysieren biochemische Reaktionen, sind Toxine zur Abwehr feindlicher Organismen, sind wichtiger Bestandteil des Immunsystems, sie bilden Körperstrukturen wie Muskeln aus und sind Transmittermoleküle.
Kohlenhydrate
BearbeitenMan unterscheidet innerhalb der Kohlenhydrate zwischen Monosacchariden, Oligosacchariden und Polysacchariden nach folgendem Schema:
| Name | Anzahl der Monosaccharid- Einheiten |
Beispiele |
|---|---|---|
| Monosaccharide | 1 | Glucose, Fructose |
| Oligosaccharide | 2–9 | Saccharose, Maltose, Raffinose |
| Polysaccharide | > 10 | Stärke, Cellulose |
Bei den Polysacchariden unterscheidet man zwischen Homopolysacchariden wie Stärke, die aus einer alternierenden Zuckereinheit aufgebaut ist, und Hetereopolysacchariden, die verschiedene Zucker enthalten.
Kohlenhydrate haben vielfältige Funktionen im Organismus. Sie sind ein Energiespeicher, welcher sehr schnell mobilisiert werden kann, bilden in Form der Chitine das Exoskelett der Gliederfüßer (Arthropoden), sind als Cellulose ein wichtiger Baustein der Zellwände von Pflanzen und sind mit der Stärke ein Energiespeicher für Pflanzen und damit auch ein wichtiger Energielieferant für die tierische und menschliche Ernährung.
Monosaccharide
BearbeitenDie verbreitetsten Monosaccharide sind die Aldohexosen und -pentosen sowie deren 2-Ketovarianten. Davon hat die Glucose eine zentrale und wichtige Rolle im Kohlenhydratstoffwechsel und damit auch im Energiehaushalt der Organismen. Der Abbau von Monosacchariden zur Energiegewinnung in Form von Adenosintriphosphat (ATP) wird Glycolyse genannt. Sie findet in praktisch allen Organismen in gleicher Form statt.
Alle Monosaccharide sind chirale Verbindungen und praktisch alle natürlich vorkommenden Monosaccharide entstammen der D-Reihe. Wurde im Stoffwechsel eine Hydroxygruppe entfernt, so spricht man von Desoxy-Zuckern. Die Desoxy-Zucker sind meist Desoxylaldosen, die normalerweise glycosidisch gebunden vorkommen. Ein Beispiel dafür ist die Desoxyribose als Baustein der Desoxyribonukleinsäure (DNA). Verzweigte Desoxyzucker werden auch als Methylosen bezeichnet und haben Bedeutung als Blutgruppensubstanz oder in den Herzglycosiden. Sie werden im Organismus durch Kohlenstoffübertragung oder Umlagerungsreaktion gebildet.[38]
Neben den Monosacchariden, die außer Sauerstoff kein weiteres Heteroatom aufweisen, sind die Aminozucker von Bedeutung. Sie sind, glycosidisch gebunden, Bestandteil von Antibiotika, Bestandteil des Mureins in den Zellwänden von Bakterien und Bausteine von Chitin-Panzern.
-
 Desoxyribose als Beispiel eines Desoxy-Zuckers
Desoxyribose als Beispiel eines Desoxy-Zuckers -
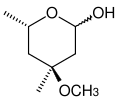 Cladinose, ein Beispiel einer Methylose
Cladinose, ein Beispiel einer Methylose


Monosaccharide kommen in der Natur frei, aber auch gebunden als Kohlenhydrate, als Zuckeranteil eines Glycosides und als Ester von anorganischen Säuren wie Phosphorsäure oder Monoschwefelsäure vor.
Sie werden im Calvinzyklus des Photosyntheseprozesses aus Kohlendioxid und Wasser in Pflanzen aufgebaut. Tiere und der Mensch müssen im Fall einer mangelnden Zufuhr von Kohlenhydraten auf Aminosäuren zurückgreifen, um hieraus Monosaccharide zu synthetisieren. Dieser Vorgang ist jedoch mit einem erhöhten Energieaufwand verbunden. Die verschiedenen Monosaccharide können von allen Organismen ineinander umgewandelt werden, so dass im Gegensatz zu den Fettsäuren und Aminosäuren keine essentiellen Zucker bekannt sind.[39]
Cyclitole
Bearbeiten
Eng verwandt mit den Monosacchariden sind die Cyclitole. Man versteht darunter Cycloalkane mit mindestens drei Hydroxygruppen. Die verbreitetsten Vertreter sind hier die Hexahydroxycyclohexane, die auch Inosite genannt werden. Sie kommen in freier Form oder phosphoryliert vor.[40] In neuerer Zeit wurde ihre Rolle als sekundärer Botenstoff (second messenger) erkannt. Durch die Substitution einer oder mehrerer Hydroxygruppen durch eine Aminogruppe erhält man Aminodesoxyinosite.
Di- und Oligosaccharide
BearbeitenOligosaccharide sind aus zwei oder mehr Zuckereinheiten aufgebaut und werden entsprechend als Di-, Tri-, Tetrasaccharide usw. bezeichnet. Das bei weitem häufigste Disaccharid ist die Saccharose (Rohr- oder Rübenzucker), welche aus einer Glucose- und einer Fructose-Einheit besteht.
Saccharose kommt in vielen Pflanzen vor. Industriell wird sie aus Zuchtformen des Zuckerrohrs (Saccharum officinarum, 14–20 % Gehalt) und der Zuckerrübe (Beta vulgaris, 16–20 % Gehalt) gewonnen.
Ein weiteres sehr wichtiges Disaccharid ist die Lactose, die in der Ernährung Neugeborener von Säugetieren (Mammalia) praktisch die einzige Kohlenhydratquelle darstellt. Lactose besteht aus 1,4-verknüpfter Galactose mit Glucose. Weitere wichtige Vertreter der Disaccharide sind Trehalose (Insekten, Pilze, Hefen, Algen, Bakterien und Moose), Gentiobiose (bspw. als Zuckerrest des Amygdalins – das Glycosid der Bittermandel (Prunus amygdalus amara)) und das Primaverin (aus Primeln (Primula)).[41]
-
 Saccharose
Saccharose -
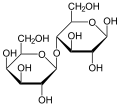 Lactose
Lactose


Polysaccharide
BearbeitenPolysaccharide sind allgegenwärtige Naturstoffe. Ein bedeutendes Polysaccharid ist die Stärke, ein pflanzlicher Reservestoff, der eine große Bedeutung für die menschliche und tierische Ernährung hat. Polysaccharide dienen als Reservestoff oder sind Strukturbildner von Zellen oder Organismen. So bilden sie auch die Grundlage für die Zellwände, die die Zellen von Bakterien und Pflanzen umgeben. Als Zellwandbestandteil der Pflanzen ist Cellulose von herausragender Bedeutung, welcher auch eine wichtige Funktion bei der Ernährung der Wiederkäuer zukommt. Weitere wichtige Polysaccharide, die in Pflanzen als Zellwandbaustein dienen, sind Pektine und Hemicellulosen. Polysaccharide, welche Aminozucker enthalten, kommen bei Tieren und Pilzen in Form von Chitin vor, welches ein Homopolysaccharid vom N-Acetyl-glucosamin darstellt.
Eine Reihe von Polysacchariden besitzen nativ oder chemisch modifiziert Bedeutung als Zusätze in der Lebensmittel-, pharmazeutischen, Textil- und Kosmetikindustrie. Sie werden entweder aus pflanzlichem Material oder biotechnologisch gewonnen. Dazu gehören Xanthan, Dextran, Levan und Pullulan.[42]
-
 Cellulose
Cellulose -
 Chitin
Chitin


Glycoside
BearbeitenGlycoside sind Konjugate von Mono- oder Oligosacchariden mit Alkoholen, Thiolen, Aldehyden oder Amiden, welche über das anomere Kohlenstoff-Atom direkt oder über ein Heteroatom verknüpft sind. Man kennt auch C-Glykoside, bei denen eine reine Kohlenstoff-Kohlenstoff-Bindung vorliegt, weil die anomere Hydroxygruppe vor der Verknüpfung entfernt wurde. Je nach Bindungstyp spricht man von O-, S-, N- oder C-Glycosiden.
Streng genommen sind Oligo- und Polysaccharide auch Glycoside, aber der Begriff Glycosid wird in der Regel nur für Konjugate mit Nicht-Kohlenhydratresten benutzt. Dieser Rest wird als Aglycon bezeichnet. Für das Aglycon wurde eine Vielzahl von Verbindungen gefunden. Hier werden nur einige wichtige Vertreter exemplarisch aufgeführt.[43]
| Funktionelle Gruppe des Aglycons mit Bindung zum Zucker | Name | Struktur | Vorkommen |
|---|---|---|---|
| Phenol | Arbutin |  |
Arbutin ist ein einfaches Glycosid und kommt in verschiedensten Früchten vor. Das Aglycon ist hier ein Hydrochinon und der Zuckerrest eine β-Glucose. |
| Alkohol | Oleandrin |  |
Oleandrin ist ein Inhaltsstoff des Oleanders (Nerium oleander) und hat als Aglycon ein Steroid |
| Alkohol | Digitoxin |  |
Digitoxin ist ein Inhaltsstoff des Roten Fingerhutes (Digitalis purpurea) und hat als Aglycon ein Steroid |
| Thiocarbonsäureamid | Sinigrin |  |
Sinigrin ist ein Inhaltsstoff des Schwarzen Senfs (Brassica nigra) und des Meerrettichs (Armoracia rusticana) und trägt als Aglycon ein Allylthiocarbonsäureamid |
| Aldehyd (Cyanhydrin) | Amygdalin |  |
Amygdalin ist das cyanogene Glycosid der Bittermandel (Prunus amygdalus amara) und trägt das Cyanhydrin von Benzaldehyd als Aglycon |
| Stickstoffheterocyclus | Adenosin |  |
Adenosin ist ein Baustein der DNA und hat als Aglycon eine Purinbase |
| Anthrachinon | Barbaloin |  |
Barbaloin kommt unter anderen in verschiedenen Aloe-Arten (Aloe) vor und trägt als Aglycon ein Anthrachinonderivat |
Glycoside finden pharmazeutisch vor allem als Herzglycoside (Digitoxin) oder als Antibiotikum (Erythromycin) Verwendung und werden aus natürlichen Quellen gewonnen. Biologisch sind sie als Bausteine der DNA und RNA von unverzichtbarer Bedeutung.
Die Bildung von Glycosiden dient in Organismen häufig dazu, ein eher apolares Aglykon in eine wasserlösliche Form zu bringen.
Peptidoglycane
BearbeitenPeptidoglycane, auch Murein genannt, sind Konjugate von Polysacchariden mit Peptiden. Sie geben den Zellwände von Bakterien ihre Festigkeit. Sie bestehen aus einem Disaccharid (N-Acetylglucosamin β-(1,4)-verknüpft mit der N-Acetylmuraminsäure), welches ein Polysaccharid bildet und über kurze Peptidketten quervernetzt ist. Die Vernetzung wird durch eine Transpeptidase gebildet, welche durch Antibiotika gehemmt werden kann und damit den Aufbau stabiler Zellmembranen verhindern kann.[44]
Lipide
BearbeitenLipide ist eine Sammelbezeichnung für unpolare und mit unpolaren organischen Lösungsmitteln wie Ether, Petrolether oder Chloroform aus organischem Material extrahierbare Verbindungen. Diese Bezeichnung ist rein historisch begründet, da hier Verbindungen extrahiert werden, die strukturell keine Ähnlichkeit zueinander haben (wie etwa Terpene oder Steroide), andere dagegen ähneln strukturell den Fetten – wie die Glycolipide.
Heute versteht man unter Lipiden solche Verbindungen, die sich von den Fetten ableiten, also Ester von Fettsäuren mit ein- oder mehrwertigen Alkoholen sind.[45]
Fettsäuren
Bearbeiten
Die häufigsten natürlichen Fettsäuren sind langkettige Carbonsäuren mit einer geradzahligen Anzahl an Kohlenstoffatomen. Man unterscheidet zwischen gesättigten und ungesättigten Fettsäuren, also ohne oder mit (einer oder mehreren) Doppelbindungen in der Alkylkette. Die Doppelbindungen von natürlichen Fettsäuren sind immer Z-konfiguriert (cis-Konfiguration).
Ein Teil der ungesättigten Fettsäuren sind für den Menschen essentiell, da sie vom Körper nicht synthetisiert werden können und daher mit der Nahrung aufgenommen werden müssen. Sie werden teilweise als Vitamin F bezeichnet.
Fettsäuren liegen in der Natur nur selten frei vor, sondern sind in der Regel über eine Esterbindung mit Alkoholen verknüpft. Die häufigste Alkoholkomponente ist Glycerin (Glycerolipide). Man kennt aber auch Ester mit Aminoalkoholen (Sphingolipide), Monosacchariden (Glycolipide), von Diolen (Diollipide) und vom myo-Inositol.[46]
Verzweigte längerkettige aliphatische Carbonsäuren werden infolge ihrer völlig verschiedenen Biosynthese nicht zu den Fettsäuren, sondern zu den Terpenen gezählt.
Eicosanoide
BearbeitenUngesättigte Fettsäuren bilden die Ausgangsverbindungen für eine Vielzahl von Regulierungsstoffen. Die Basis hierfür bildet die Arachidonsäure, welche eine ungesättigte Fettsäure ist und zwanzig Kohlenstoffatome enthält. Daher leitet sich der Name Eicosanoide ab. Die Arachidonsäure wird im Organismus aus der essentiellen Linolsäure durch Kettenverlängerung und Dehydrierung gebildet.

Das Gerüst der Prostaglandine, die die wichtigsten Eicosanoide darstellen, leitet sich von der Prostansäure ab. Sie bestehen immer aus einem Fünfring mit zwei zueinander benachbarten Seitenketten. Sowohl die Seitenketten als auch der Fünfring können verschiedene funktionelle Gruppen tragen.
Die analogen Verbindungen mit einem Sechsring werden als Thromboxane bezeichnet.
Fette
Bearbeiten
Fette sind die Ester von Fettsäuren mit Glycerin. Dabei handelt es sich meist um Triglyceride, denn Mono- und Diglyceride spielen nur als Stoffwechselintermediat eine Rolle und kommen selten frei vor. Da Glycerin ein dreiwertiger Alkohol ist, findet man neben Estern von drei Molekülen der gleichen Fettsäure auch gemischte Ester. Wenn der Glycerinrest dadurch ein Asymmetriezentrum bekommt, sind Fette chirale Verbindungen und durch die Biosynthese über das L-Glycerin-3-phosphat (G3P) optisch aktiv.
Fette kommen in allen Tieren als Energiespeicher in spezialisierten Geweben vor, adipöses Gewebe kann bis zu 80 % Fett enthalten. Auch die Samen verschiedener Pflanzen speichern Fette.
Eine Trennung verschiedener Fette ist normalerweise nicht möglich, da es sich um multiple Stoffgemische handelt, die sehr ähnliche chemische und physikalische Eigenschaften aufweisen.
Wachse
Bearbeiten
Wachse sind unpolare Ester von Fettsäuren und cyclischen oder langkettigen aliphatischen Alkoholen. Natürlich vorkommende Wachse sind in der Regel schwer trennbare Stoffgemische, welche eher von technischer Bedeutung sind. Wachse dienen in der Regel als Strukturbildner wie etwa bei den Bienenwaben und können meist wahrscheinlich nicht mehr dem Stoffwechsel zugeführt werden. Eine Ausnahme bilden bestimmte Meerestiere, die Wachse als Reservestoffe herstellen, wie z. B. Walrat (Cetaceum). Bei Pflanzen bilden Wachse die Cuticula als Verdunstungsschutz. Der Alkylrest der Alkoholgruppe kann verzweigt oder unverzweigt sein. Bei Säugetieren besteht die Alkoholgruppe meist aus Cholesterin.[47]
Komplexe Lipide
BearbeitenAls komplexe Lipide oder membranbildende Lipide werden solche Lipide bezeichnet, die am Aufbau von Zellmembranen beteiligt sind. Diese Lipide tragen außer den unpolaren Fettsäureresten polare Gruppen. Durch diese polaren Gruppen erhalten sie die Fähigkeit zur Selbstorganisation in wässrigen Medien. Dies lässt sich unter Laborbedingungen durch die Bildung von Liposomen zeigen.[48] Einen ähnlichen Aufbau findet man in Zellmembranen. In wässrigen Medien organisieren sich die polaren Gruppen in Richtung des polaren Wassers und die unpolaren Gruppen bilden eine Lipiddoppelschicht aus – mit Wasser außerhalb und innerhalb der Lipiddoppelschicht.[49]


Phospholipide, Sphingolipide und Glycolipide sind zellmembranbildende Lipide. Sie unterscheiden sich durch ihre polaren Reste. Im Fall der Phospholipide handelt es sich um einen Phosphorsäureester des Diglycerides. Am Phosphatrest befinden sich noch polare Reste wie Cholin (Lecitin) oder Ethanolamin (Kephalin). Sphingolipide leiten sich im Gegensatz zu den Phospholipen vom Sphingosin ab. Über eine Amidbindung ist die Fettsäure an das Sphingosin gebunden, das wiederum über einen Phosphatrest durch Esterbindungen mit einer polaren Gruppe wie Serin, Ethanolamin oder Cholin verbunden ist. Glycolipide hingegen sind verschiedene Fettsäurederivate mit einer Zuckergruppe als polarem Rest. Diese können entweder vom Glycerid-Typ, vom Sphingolipid-Typ oder einfache Fettsäureester der Monosaccharide sein.[50]
Isoprenoide Verbindungen
BearbeitenIsoprenoide Verbindungen leiten sich vom Isopren ab und sind formal Oligomere oder Polymere des Isoprens. Dieses Prinzip wurde vom Nobelpreisträger Leopold Ružička erkannt.[51] Daraus wird die Naturstoff-Gruppe der Terpene und Steroide gebildet. Letztere sind im eigentlichen Sinne ebenfalls Terpene, werden aber aufgrund ihrer besonderen biologischen Bedeutung separat betrachtet. Allen Terpenen ist gemeinsam, dass sie aus der Mevalonsäure und über den gleichnamigen Mevalonsäureweg aufgebaut werden.
Terpene
BearbeitenDie Stoffgruppe der Terpene besitzt eine riesige Vielfalt an Kohlenstoff-Gerüsten. Allen gemeinsam ist jedoch, dass sie sich vom Isopren ableiten und Vielfache dieses Moleküls darstellen. Man unterscheidet die Terpene nach der Anzahl ihrer Kohlenstoffatome. Praktisch alle Terpengerüste tragen Trivialnamen und sind nach ihrer biologischen Quelle benannt. Funktionelle Gruppen werden dabei häufig als Präfix oder Suffix zum Namen des Kohlenstoffgerüstes hinzugefügt.
| Name | Anzahl der Kohlenstoffatome | Anzahl der Isopreneinheiten | Beispiele |
|---|---|---|---|
| Isopren | 5 | 1 | Isopren |
| Monoterpene | 10 | 2 | Menthol, Carvon, Thujanon, Campher |
| Sesquiterpene | 15 | 3 | Farnesol, Sesquisabinen, Cadalenol, Artemisiten |
| Diterpene | 20 | 4 | Retinal (Vitamin A), Paclitaxel, Rosan, Nimbion, Gibberellan |
| Sesterterpene | 25 | 5 | Neomanoalid, Scalarin, Hyrial |
| Triterpene | 30 | 6 | Squalen, Protostan, Lanosterol, Oleanan |
| Tetraterpene | 40 | 8 | Carotine (Provitamin A), Xanthophylle |
| Polyterpene | >40 | >8 | Naturkautschuk, Guttapercha |
Zu den Monoterpenen zählen auch die Iridoide, welche sich durch den Grundkörper Iridodial auszeichnen.[52]

Die Sesterterpene stellen eine quantitativ kleine Gruppe an Terpenen dar. Wenn während des Stoffwechsels Kohlenstoffatome entfernt werden, dann bekommen die Verbindungen das Präfix Nor wie etwa das Sesquiterpen Norpatchoulenol, bei welchem aus dem ursprünglichen Patchoulialkohol formal ein Methan-Molekül entfernt wurde.
Terpene erfüllen eine Vielzahl von biologischen Funktionen, die von Aromen und Duftstoffen über Pheromone bis zu Vitaminfunktionen (Vitamin A) und Hormonvorläufern (Steroidhormone) reichen.[53] Ihre technischen Anwendungen reichen von Pharmaka (Taxol) oder Steroiden, über Insektizide (Pyrethroide) bis zu den Geruchsstoffen für die Kosmetikindustrie.
Steroide
BearbeitenSteroide sind in der Tier- und Pflanzenwelt weit verbreitete Naturstoffe. Sie leiten sich alle vom Triterpen Squalen ab, welches zum tetracyclischen Steran-Gerüst cyclisiert. Bei den natürlich vorkommenden Steroiden sind die Ringe B und C sowie die Ringe C und D jeweils trans verbunden und diese werden Gonane genannt. Die Ringe A und B des Gonans können entweder cis-(5β-Gonan) oder trans-(5α-Gonan) verbunden sein. Bei den natürlichen Steroiden sind diese stets cis-verbunden, also 5β-Gonane. Das wichtigste Steroid bei Menschen und Tieren ist das Cholesterin, welches in Pflanzen nicht vorhanden ist. Aus Cholesterin werden Lipoproteine und Steroidhormone aufgebaut, wie die Hormone der Nebennierenrinde (Corticosteroide). Die Sexualhormone von Säugetieren, also auch von Menschen, sind Steroide.[54]
-
 Strukturformel von Steran.
Strukturformel von Steran. -
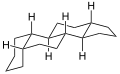 5β-Gonan
5β-Gonan


Polyprenylhydrochinone
BearbeitenDas Vitamin K des Menschen sowie mehrere andere ähnliche Stoffe in allen Lebewesen haben die Gemeinsamkeit eines Hydrochinonrests sowie einen angehängten Polyprenylrest. Bekannteste Beispiele und ihre Abkürzungen sind Ubichinone (beim Menschen Ubichinon-10, Coenzym Q-10), Phyllochinon (Vitamin K), Menachinone (MK), Plastochinon (PQ) und Tocochinon. Zum sicheren Nachweis dieser Substanzen sind relativ aufwändige Probenvorbereitungen und Analysenverfahren erforderlich, wie z. B. beim Tocochinon.[55][56][57][58][59][60] Sie fungieren als Elektronentransporter in der mitochondriellen und bakteriellen Atmungskette.
-
 Coenzym Q-10
Coenzym Q-10 -
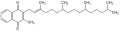 Phyllochinon
Phyllochinon -
 Menachinon Grundstruktur
Menachinon Grundstruktur -
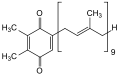 Plastochinon
Plastochinon



Aromatische Verbindungen
BearbeitenIn Organismen kommen grundsätzlich drei Biosynthesewege vor, die zu aromatischen Verbindungen führen – der Shikimatweg, der Malonatweg und der Mevalonatweg.[61]
Der Shikimatweg beruht auf dem Kohlenhydratstoffwechsel und verläuft über die Shikimisäure zu den aromatischen Naturstoffen. Dieser Weg findet vor allem in höheren Pflanzen statt. Charakteristisch für diese Naturstoffe sind häufig hoch oxidierte phenolische Aromaten mit einer linearen Seitenkette mit funktionellen Gruppen. Die phenolischen Gruppen befinden sich meist in 3-, 4- und 5-Stellung der Seitenkette.

Der Malonatweg (Polyketidweg) beruht auf dem Fettsäurestoffwechsel. Durch Aneinanderlagerung von Acetat-Einheiten entsteht eine Polycarbonylverbindung, die in einer oder mehreren Aldolkondensationen zu ein- oder mehrkernigen Aromaten cyclisieren kann. Charakteristisch für diese Verbindungen sind hoch oxidierte Aromaten, die häufig auch Chinone oder Hydrochinone enthalten, jedoch ohne längere Seitenketten. Die Sauerstoffgruppen stehen also in 1,4-Stellung. Den Malonatweg findet man vor allem in Mikroorganismen.

Auch der Mevalonatweg führt zu aromatischen Naturstoffen, den Terpenen. Ein Beispiel ist die Biosynthese von Thymol. Die auf diesem Weg hergestellten Aromaten tragen häufig die für Terpene charakteristischen Isopropylgruppen.

Tierische Organismen stellen aromatische Verbindungen selten selbst her. Diese sind daher normalerweise essentielle Nahrungsbestandteile (aromatische Aminosäuren und Vitamine).[63]
Phenylpropan-Derivate
BearbeitenPhenylpropan-Derivate sind aromatische Verbindungen mit einer Propyl-Seitenkette. Der Aromat trägt dabei häufig Hydroxy- oder Methoxy-Gruppen. Die Propyl-Seitenkette kann sowohl gesättigt oder ungesättigt sein, einen Cyclus bilden oder verschiedene funktionelle Gruppen tragen. Diese Verbindungsklasse wird in Pflanzen und Mikroorganismen über den Shikimat-Biosyntheseweg gebildet. Phenylpropanoide bilden neben den Terpenen den zweithäufigsten Bestandteil der ätherischen Öle. Bekannte Phenylpropane sind der Zimtaldehyd, das Anethol und das Estragol. Die Lignine sind als Baustoff der Hölzer ein Polymer der Phenylpropan-Derivate.
-
 Coniferylalkohol
Coniferylalkohol -
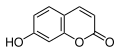 Umbelliferon
Umbelliferon

Flavonoide
Bearbeiten
Flavonoide gehören zu den Pflanzenfarbstoffen und leiten sich strukturell von den Phenylpropanoiden ab. Daher findet man hier auch häufig Phenole oder Methoxyphenole. Sie sind häufig mit Kohlenhydraten glycosidisch gebunden und bilden das Aglycon. Je nach funktioneller Gruppe am heterocyclischen Ring unterscheidet man zwischen Flavan, Flavon, Flavonol, Flavonon und Flavonolol. Flavonoide sind vor allem als Pflanzenfarbstoffe von Bedeutung und bilden die Mehrzahl aller Blütenfarbstoffe.[64]
Gerbstoffe
BearbeitenUnter dem Oberbegriff Gerbstoffe werden anorganische und organische Verbindungen bezeichnet, die in der Lage sind, tierische Häute in Leder zu überführen. Den organischen Gerbstoffen ist gemeinsam, dass sie phenolische Gruppen enthalten, aber sie bilden keine einheitliche Stoffklasse. Der wichtigste und bekannteste Gerbstoff, welcher zu den Naturstoffen zählt, ist das Tannin und ist ein Polyhydroxyphenol. Zu den Gerbstoffen zählen auch einige phenolische Flavone und deren dimere Kondensationsprodukte. Häufig liegen diese, wie auch die Flavone selbst, als Aglycon von Glycosiden vor.[65]
Polyketide
BearbeitenDie Polyketide sind eine große und sehr heterogene Naturstoffgruppe. Sie umfasst sowohl aliphatische, cyclische, acyclische und aromatische Verbindungen. Ihre biologischen Funktionen sind häufig unbekannt. Sie haben sehr große strukturelle Unterschiede, aber gehören alle der gleichen Naturstoffklasse an. Polyketide zeichnen sich durch einen gemeinsamen Biosyntheseweg aus. Allen ist gemeinsam, dass sie ein Kohlenstoffrückgrat enthalten, welches aus Essigsäure und Propionsäure aufgebaut ist. Wie bei den Terpenen unterscheidet man bei den Polyketiden nach Anzahl der Acetateinheiten.
| Name | Anzahl der Acetate | Beispiele |
|---|---|---|
| Triketide | 3 | 
|
| Tetraketide | 4 | 
|
| Pentaketide | 5 | 
|
| Heptaketide | 7 | 
|
| Polyketidalkaloide | Einbau von Ammoniak | 
|
| Anthrachinone | 8 | 
|
| Tetracycline | 8 Malonateinheiten | 
|
Polyketocarbonsäuren bilden den Ausgang aller Ketide. Grundsätzlich findet man in der Biosynthese von Polyketiden die Reaktionen der Claisen-Esterkondensation, der Aldolkondensation und der Dieckmann-Kondensation.

Als Folgereaktionen kennt man Aldolreaktionen, die Bildung von Enolestern oder -ethern, Methylierungen, Chlorierungen oder Hydroxylierungen, aber auch Reduktionen der Carboxyl- oder Carbonylgruppen zu Alkoholen bzw. Methylengruppen. Auch die Decarboxylierung der β-Ketosäure-Gruppe wird beobachtet.
Heterozyklen
BearbeitenPteridine
Bearbeiten
Die Pteridine leiten sich von der Grundstruktur des Pteridin ab, so beispielsweise die Vitamine Riboflavin (Vitamin B2) und Folsäure, die Coenzyme FAD und FMN[66] sowie die Molybdän-Cofaktoren, die sich alle vom Molybdopterin ableiten. Ihre Funktionen als Cofaktoren sind vielfältig.
-
 Riboflavin
Riboflavin -
 Folsäure
Folsäure -
 FAD
FAD -
 Molybdän-Cofaktor
Molybdän-Cofaktor



Pyridinderivate
BearbeitenInsbesondere Nicotinsäure (Vitamin B3) und Pyridoxalphosphat mit seinen Vorstufen Pyridoxin, Pyridoxal und Pyridoxamin (Vitamin B6) fallen in diese Substanzklasse.[67]
-
 Nicotinsäure
Nicotinsäure -
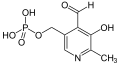 Pyridoxalphosphat
Pyridoxalphosphat -
 Pyridoxin
Pyridoxin -
 Pyridoxamin
Pyridoxamin




Nucleoside
BearbeitenAls Nucleoside werden N-Glycoside von heterocyclischen Systemen bezeichnet. Im engeren Sinn werden besonders die Bausteine der DNA und RNA als Nucleinsäuren so bezeichnet. Bei diesen Nucleinsäuren ist der Zuckerrest immer eine Ribose (RNA) oder Desoxyribose (DNA). In der DNA sind mit Hilfe der Desoxyribonucleinsäuren die Erbinformationen gespeichert. Die RNA kann mit Hilfe der Ribonukleinsäuren biochemische Reaktionen katalysieren und als Signalüberträger bzw. als Informationsspeicher dienen.
Man kennt natürliche Nucleoside mit Purinbasen (Adenin, Guanin) und mit Pyrimidinbasen (Cytosin, Thymin und Uracil).[68]

| Purine | Pyrimidine | ||
|---|---|---|---|
 Adenin (A) |
 Cytosin (C) | ||
 Guanin (G) |
| ||
| Strukturformeln der Nukleobasen in DNA (A,G,C,T) und RNA (A,G,C,U). Die N-glycosidische Bindung zu Ribose oder Desoxyribose in DNA findet jeweils an der in den Abbildungen nach unten zeigenden NH-Gruppe statt. | |||


Porphyrine und Corrinoide
Bearbeiten
Zu den primären Naturstoffen zählen noch eine Reihe weiterer Verbindungsklassen wie die Tetrapyrrole, welche aus vier Pyrrol-Resten, die über eine Methin-Brücke verbunden sind, gebildet werden. Die größte Bedeutung hier haben die ringförmigen Tetrapyrrole, die Porphyrine und Chlorine. Die Phorphyrine bilden die Grundlage für das Chlorophyll, Cytochrom und Hämoglobin und sind der Komplexligand für ein Eisen(II)-Atom. Die Chlorine sind in den Chlorophyllen der Komplexligand für Magnesium(II) als Zentralatom. Sie haben vielfältige Aufgaben im Organismus, die von Sauerstofftransport bzw. -speicherung (Hämoglobin und Myoglobin) über den Elektronen- und Energietransfer bis zur Katalyse biochemischer Reaktionen (Vitamin B12 und Cytochrom P 450) als Coenzym reichen.[69]
Alkaloide
BearbeitenBereits im Jahre 1806 wurde Morphin vom deutschen Apotheker Friedrich Sertürner als erstes Alkaloid isoliert. Der Begriff Alkaloide wurde 1819 von Carl Friedrich Wilhelm Meißner geprägt, der darunter alle basischen Naturstoffe verstand. Später wurde der Begriff auf andere stickstoffhaltige Naturstoffe erweitert. Heute fasst man unter dieser Bezeichnung alle stickstoffhaltigen Naturstoffe zusammen, auch wenn es bis heute keine einheitliche Definition gibt. Alkaloide haben oft biologische Wirkungen und bilden wichtige Grundlagen als Leitstrukturen für pharmazeutische Wirkstoffe.
Es gibt verschiedene Bezeichnungen für die Alkaloid-Klassen, die in der Literatur nicht einheitlich gehandhabt werden. Zum einen werden Alkaloide nach ihrer botanischen Herkunft – Solanum-, Papaver-, Angostura-, Lobelia-Alkaloide usw. bezeichnet – aber zum anderen auch durch ihre chemische Stammverbindung in Pyridin-, Chinolin- oder Steroid-Alkaloide unterteilt.
Häufig werden als Alkaloide nur Verbindungen bezeichnet, die sich von den proteinogenen Aminosäuren ableiten und aromatische Stickstoff-Heterocyclen enthalten. Nach dieser Definition allerdings sind verschiedene stickstoffhaltige Naturstoffe wie Coniin, Piperin und Coffein keine Alkaloide.
Auch die systematische Einteilung der Alkaloide ist nicht einheitlich. Zum einen gibt es die Einteilung nach ihrer chemischen Struktur, also nach der Art des Stickstoff-Heterocyclus: So gibt es dann beispielsweise Steroid-, Indol-, Pyridin- oder Tropan-Alkaloide. Auch die Einteilung nach dem Ursprung ist verbreitet: Mutterkorn-Alkaloide, Curare oder Opiate.
In der heutigen chemischen Literatur[70][71] werden Alkaloide in folgende Gruppen, die nach ihrer chemischen Struktur geordnet sind, zusammengefasst:
- Alkaloide mit Piperidin-, Pyrrol-, Pyrrolidin- und Pyridin-Gerüst
- Alkaloide mit Isochinolin-, Chinolin-, Chinazolin- und Indol-Gerüst
- Alkaloide mit Indolizin, Pyrrolizidin- und Chinolizidin-Gerüst
- Purin-Alkaloide
- Steroid-Alkaloide
Biogene Amine
BearbeitenBiogene Amine sind Verbindungen, die durch einfaches Decarboxylieren einer Aminosäure gebildet werden und als wichtiger Bestandteil von Lipiden, als Coenzym oder als Neurotransmitter (Acetylcholin, Tryptamin, Serotonin oder Histamin) eine Rolle spielen. Pharmazeutisch spielt hier vor allem das L-Dopa als Parkinson-Medikament eine wichtige Rolle. Weitere bekannte Vertreter sind das Adrenalin, Ephedrin oder Mescalin.

Siehe auch
BearbeitenLiteratur
Bearbeiten- Gerhard Habermehl, Peter E. Hammann, Hans C. Krebs, W. Ternes: Naturstoffchemie, 3., vollst. überarb. und erw. Auflage, Springer Verlag 2008, ISBN 978-3-540-73732-2.
- Peter Nuhn: Naturstoffchemie. Mikrobielle, pflanzliche und tierische Naturstoffe, 4. Auflage, S. Hirzel Verlag, Stuttgart 2006, ISBN 978-3-7776-1363-5.
Weblinks
BearbeitenEinzelnachweise
Bearbeiten- ↑ Peter Nuhn: Naturstoffchemie. Mikrobielle, pflanzliche und tierische Naturstoffe, 2. Auflage, S. Hirzel Verlag, Stuttgart 1990, S. 20–23; ISBN 3-7776-0473-9.
- ↑ Otto Krätz: 7000 Jahre Chemie, Nikol Verlagsgesellschaft, Hamburg 1999; ISBN 3-933203-20-1.
- ↑ Erklärung der Begriffe und Abkürzungen. In: REACH Helpdesk. 24. Juni 2013, abgerufen am 29. Juli 2019.
- ↑ a b Verordnung (EG) Nr. 1907/2006 zur Registrierung, Bewertung, Zulassung und Beschränkung chemischer Stoffe (REACH) in der konsolidierten Fassung vom 2. Juli 2019
- ↑ BMAS – Gesetze: Biostoffverordnung ( vom 15. August 2014 im Internet Archive)
- ↑ A. Fleming: On the antibacterial action of cultures of a penicillium, with special reference to their use in the isolation of B. influenzae. 1929. In: Bull. World Health Organ.. Band 79, Nummer 8, 2001, S. 780–790, PMID 11545337. PMC 2566493 (freier Volltext).
- ↑ Mansukhlal C. Wani, Harold Lawrence Taylor, Monroe E. Wall, Philip Coggon, Andrew T. McPhail: Plant antitumor agents. VI. Isolation and structure of taxol, a novel antileukemic and antitumor agent from Taxus brevifolia, in: J. Am. Chem. Soc., 1971, 93, S. 2325–2327; doi:10.1021/ja00738a045.
- ↑ Gerhard Höfle, Norbert Bedorf, Heinrich Steinmetz, Dietmar Schomburg, Klaus Gerth, Hans Reichenbach: Epothilon A und B – neuartige, 16gliedrige Makrolide mit cytotoxischer Wirkung: Isolierung, Struktur im Kristall und Konformation in Lösung, in: Angewandte Chemie, 1996, 108, S. 1671–1673; doi:10.1002/ange.19961081342.
- ↑ Neueinfuehrung Zidovudin. In: Zeitschrift für Chemotherapie, 1987, Heft 4. Abgerufen am 4. August 2013.
- ↑ Lee Jia, Yuqing Zhao: Current Evaluation of the Millennium Phytomedicine – Ginseng (I): Etymology, Pharmacognosy, Phytochemistry, Market and Regulations, in: Curr. Med. Chem., 2009, 16, S. 2475–2484; PMC 2752963 (freier Volltext).
- ↑ Lee Jia, Yuqing Zhao, Xing-Jie Liang: Current Evaluation of the Millennium Phytomedicine – Ginseng (II): Collected Chemical Entities, Modern Pharmacology, and Clinical Applications Emanated from Traditional Chinese Medicine, in: Curr. Med. Chem., 2009, 16, S. 2924–2942; PMC 2754208 (freier Volltext).
- ↑ Steven D. Ehrlich: Ginkgo Biloba Review. University of Maryland – Medical Center, 13. Dezember 2010, abgerufen am 4. August 2013.
- ↑ S. Ganguli: Neem: A therapeutic for all seasons ( vom 5. Juni 2011 im Internet Archive) (PDF; 21 kB), in: Current Science, 2002, 82, S. 1304 (Archive.org).
- ↑ Bernd Schäfer: Naturstoffe der chemischen Industrie, Spektrum Akademischer Verlag, Heidelberg 2006; ISBN 978-3-8274-1614-8.
- ↑ Gareth Thomas: Medicinal Chemistry, 2. Auflage, John Wiley & Sons Ltd, West Sussex, 2007, S. 90–110, 161–163; ISBN 978-0-470-02598-7.
- ↑ Rolf Breinbauer, Ingrid R. Vetter, Herbert Waldmann: Von Proteindomänen zu Wirkstoffkandidaten – Naturstoffe als Leitstrukturen für das Design und die Synthese von Substanzbibliotheken, in: Angewandte Chemie, 2002, 114, S. 3002–3015; doi:10.1002/1521-3757(20020816)114:16<3002::AID-ANGE3002>3.0.CO;2-V.
- ↑ Herbert Waldmann: Natürlich kombinatorisch – naturstoffgetriebene Wirkstoffentwicklung, in: Nachrichten aus der Chemie, 2003, 51, S. 126–131; doi:10.1002/nadc.20030510210.
- ↑ Rudolf Hänsel, Otto Sticher (Hrsg.): Pharmakognosie – Phytopharmazie. 9. Auflage. Springer Verlag, Heidelberg 2009, ISBN 978-3-642-00962-4, S. 18–29.
- ↑ W. Kreis: Prinzipien des Sekundärstoffwechsels. (PDF) In: Phytochemische Grundlagen. SWBplus Kataloganreicherungen, abgerufen am 5. August 2013.
- ↑ Wolfgang Kraus, Michael Bokel, Adolf Klenk, Helmut Pöhn: The structure of azadirachtin and 22,23-dihydro-23β-methoxyazadirachtin, in: Tetrahedron Letters, 1985, 26, S. 6435–6438; doi:10.1016/S0040-4039(00)99020-8.
- ↑ G. Büchi, R. E. Erickson, N. Wakabayashi: Terpenes. XVI.1,2 Constitution of Patchouli Alcohol and Absolute Configuration of Cedrene, in: J. Am. Chem. Soc., 1961, 83, S. 927–938; doi:10.1021/ja01465a042.
- ↑ G. Büchi, William D. Macleod: Synthesis of Patchouli Alcohol, in: J. Am. Chem. Soc., 1962, 84, S. 3205–3206; doi:10.1021/ja00875a047.
- ↑ Gareth Thomas: Medicinal Chemistry S. 90–110.
- ↑ Peter Bützer: Aufsatz über Vitamin B12. (PDF; 481 kB) In: Molekulare Dynamik – Systemdynamik. Januar 2008, archiviert vom (nicht mehr online verfügbar) am 6. Dezember 2012; abgerufen am 4. August 2013.
- ↑ K.C. Nicolaou, E.J. Sorensen: Classics in Total Synthesis: Targets, Strategies, Methods, VCH Verlagsgesellschaft mbH, Weinheim, 1996, S. 711–729; ISBN 3-527-29284-5.
- ↑ K.C. Nicolaou, E.J. Sorensen: Classics in Total Synthesis: Targets, Strategies, Methods, S. 655–671.
- ↑ The Nobel Prize in Chemistry 1981. In: Nobelprize.org. Abgerufen am 4. August 2013.
- ↑ The Nobel Prize in Chemistry 1965. In: Nobelprize.org. Abgerufen am 4. August 2013.
- ↑ K.C. Nicolaou, E.J. Sorensen: Classics in Total Synthesis: Targets, Strategies, Methods, S. 99–134.
- ↑ K.C. Nicolaou, S.A. Snyder: Classics in Total Synthesis II, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2003; ISBN 978-3-527-30684-8.
- ↑ Stefan Abrecht, Peter Harrington, Hans Iding, Martin Karpf, René Trussardi, Beat Wirz, Ulrich Zutter: The Synthetic Development of the Anti-Influenza Neuraminidase Inhibitor Oseltamivir Phosphate (Tamiflu®): A Challenge for Synthesis & Process Research, in: CHIMIA International Journal for Chemistry, 2004, 58, S. 621–629.
- ↑ R. Carle: Sekundäre Pflanzenstoffe – Abwehrstoffe und Nutraceuticals? In: vetline.de. 10. Oktober 2007, abgerufen am 5. August 2013. (PDF).
- ↑ Bernhard Watzl, Claus Leitzmann: Bioaktive Substanzen in Lebensmitteln. 3. Auflage. Hippokrates, Stuttgart 2005, ISBN 3-8304-5308-6, S. 15.
- ↑ Peter Nuhn: Naturstoffchemie, S. 23.
- ↑ Hans Beyer, Wolfgang Walter: Lehrbuch der organischen Chemie, 21. Auflage, S. Hirzel Verlag, Stuttgart 1988, S. 822–828; ISBN 3-7776-0438-0.
- ↑ Hans-Dieter Belitz, Walter Grosch: Lehrbuch der Lebensmittelchemie. 4. Auflage. Springer Verlag, Heidelberg/Berlin 1992, ISBN 3-540-55449-1, S. 9.
- ↑ Peter Nuhn: Naturstoffchemie, S. 77.
- ↑ Peter Nuhn: Naturstoffchemie, S. 159–160.
- ↑ Gerhard Michal: Biochemical Pathway, Spektrum Akademische Verlagsgesellschaft, Heidelberg 1999, S. 37–40; ISBN 3-86025-239-9.
- ↑ Beyer, Walter: Lehrbuch der Organischen Chemie, S. 405.
- ↑ Peter Nuhn: Naturstoffchemie, S. 200–201.
- ↑ Peter Nuhn: Naturstoffchemie, S. 209.
- ↑ Peter Nuhn: Naturstoffchemie, S. 174–181.
- ↑ Gerhard Habermehl, Peter E. Hammann, Hans C. Krebs, Naturstoffchemie, 2. Auflage, Springer Verlag, 2002, S. 385; ISBN 3-540-43952-8.
- ↑ Peter Nuhn: Naturstoffchemie, S. 297.
- ↑ Nomenclature of Glycolipids. In: IUPAC. Abgerufen am 28. Januar 2016.
- ↑ Peter Nuhn: Naturstoffchemie, S. 397.
- ↑ Roger R. C. New (Editor), Liposomes a practical approach, IRL Press at Oxford University Press, Oxford 1990, S. 13; ISBN 0-19-963077-1.
- ↑ William H. Elliott, Daphne C. Elliott: Biochemistry and Molecular Biology, forth ed., Oxford University Press, Oxford 2009, S. 16–26; ISBN 978-0-19-922671-9.
- ↑ Peter Nuhn: Naturstoffchemie, S. 311–322.
- ↑ Leopold Ružička: Nobel Lecture: Multimembered Rings, Higher Terpene Compounds and Male Sex Hormones. In: Nobelprize.org. 12. Dezember 1945, abgerufen am 6. August 2013. (PDF; 525 kB)
- ↑ Katharina Munk: Grundstudium Biologie: Botanik. Spektrum Akademischer Verlag, Heidelberg 2001; ISBN 3-8274-0909-8.
- ↑ Eberhard Breitmaier: Terpene: Aromen, Düfte, Pharmaka, Pheromone, B.G. Teubner, Stuttgart 1999; ISBN 3-519-03548-0.
- ↑ Beyer, Walter: Lehrbuch der Organischen Chemie, S. 678–679, 686–698.
- ↑ Kanazawa H, Miyata C, Nagata Y, Urano S, Matsushima Y: Determination of alpha-tocopherol and alpha-tocopherylquinone in rat tissues and plasma by high-performance liquid chromatography with electrochemical detection., Chem Pharm Bull (Tokyo). 2000 Oct;48(10):1462-6, PMID 11045451.
- ↑ H.-U. Melchert, D. Pollok D, E. Pabel, K. Rubach, H.-J. Stan: Determination of tocopherols, tocopherolquinones and tocopherolhydroquinones by gas chromatography-mass spectrometry and preseparation with lipophilic gel chromatography.: In J. Chromatogr. A. 2002 Nov 8;976(1-2):215-20, PMID 12462612
- ↑ Mottier P, Gremaud E, Guy PA, Turesky RJ: Comparison of gas chromatography-mass spectrometry and liquid chromatography-tandem mass spectrometry methods to quantify alpha-tocopherol and alpha-tocopherolquinone levels in human plasma., Anal Biochem. 2002 Feb 1;301(1):128-35, PMID 11811977.
- ↑ D. Pollok, H.-U. Melchert: Determination of alpha-tocopherolquinone in human serum samples by liquid chromatography with fluorescence detection and on-line post-column derivatization: In J. Chromatogr. A. 2004 Nov 12;1056(1-2):257-62, PMID 15595560
- ↑ Dragoun M, Klausová K, Šimicová P, Honzíková T, Stejskal J, Navrátilová K, Hajšlová J, Bárta J, Bártová V, Jarošová M, Bjelková M, Filip V, Kyselka J: Formation of Previously Undescribed Δ7-Phytosterol Oxidation Products and Tocopherylquinone Adducts in Pumpkin Seed Oil during Roasting, Screw-Pressing, and Simulated Culinary Processing at Elevated Temperatures., J Agric Food Chem. 2022 Sep 21;70(37):11689-11703, PMID 36094395.
- ↑ Shichiri M, Harada N, Ishida N, Komaba LK, Iwaki S, Hagihara Y, Niki E, Yoshida Y: Oxidative stress is involved in fatigue induced by overnight deskwork as assessed by increase in plasma tocopherylhydroqinone and hydroxycholesterol., Biol Psychol. 2013 Dec;94(3):527-33, PMID 24121154.
- ↑ Gerhard Michal: Biochemical Pathway, Spektrum Akademische Verlagsgesellschaft, Heidelberg, 1999, S. 59–60, 85–86 und 192; ISBN 3-86025-239-9.
- ↑ Mikio Yamazaki, Taeko Usui, Shoji Shibata: The Biogenesis of Plant Products. II. The Biogenesis of Thymol.; in: Chemical & Pharmaceutical Bulletin, 1963, 11, S. 363–365; doi:10.1248/cpb.11.363 (Volltext).
- ↑ Peter Nuhn: Naturstoffchemie, S. 522.
- ↑ Peter von Sengbusch, Paul von Sengbusch: Phenolische Substanzen. In: Der Sekundärstoffwechsel der Pflanzen. Sekundäre Pflanzenprodukte (Pflanzenstoffe). Botanik online 1996–2004 der Universität Hamburg, abgerufen am 6. August 2013.
- ↑ Otto Th. Schmidt, Walter Mayer: Natürliche Gerbstoffe, in: Angewandte Chemie, 1956, 68 (3), S. 103–115; doi:10.1002/ange.19560680305.
- ↑ Beyer, Walter: Lehrbuch der Organischen Chemie, S. 801–803, 882–883.
- ↑ Beyer, Walter: Lehrbuch der Organischen Chemie, S. 762, 768–769.
- ↑ Beyer, Walter: Lehrbuch der Organischen Chemie, S. 851–858.
- ↑ Beyer, Walter: Lehrbuch der Organischen Chemie, S. 710–717.
- ↑ Gerhard Habermehl, Peter E. Hammann, Hans C. Krebs: Naturstoffchemie, S. 131–243.
- ↑ Peter Nuhn: Naturstoffchemie, S. 553–597.